Abstract
Considerable advances toward understanding the molecular mechanisms of adrenocortical tumors (ACT) development have recently been made. The study of rare genetic syndromes associated with ACT has greatly facilitated progress and has increased our understanding of sporadic adrenal tumors. Furthermore, several observations have demonstrated that genetic alterations are frequent in both benign and malignant ACT. This chapter summarizes recent advances in the genetics of ACT, highlighting both improvements in our understanding of the pathophysiology and the diagnosis of these tumors.
Keywords
adrenocortical tumors (ACTs), Li–Fraumeni syndrome, Beckwith–Wiedemann syndrome, Cushing’s syndrome, Carney complex, MEN1, McCune–Albright syndrome
The identification of germline molecular defects in hereditary syndromes responsible for adrenocortical tumors (ACTs) has unraveled new pathophysiological mechanisms. Indeed, similar molecular defects have since been identified as somatic alterations in sporadic tumors. The familial diseases are Li–Fraumeni syndrome, which may be due to germline mutation of the tumor suppressor gene TP53 ; Beckwith–Wiedemann syndrome, which is caused by dysregulation of the imprinted IGF-II locus at 11p15; type 1 multiple endocrine neoplasia (MEN1), which is characterized by a germline mutation of the menin gene; and Cushing’s syndrome due to primary pigmented nodular adrenocortical disease (PPNAD), which has been observed in Carney complex patients presenting inactivating germline PRKAR1A mutations or inactivating germline PDE11A mutations. Interestingly, allelic losses at 17p13 and 11p15 have been demonstrated in sporadic adrenocortical cancer, and somatic PRKAR1A mutations have been found in secreting adrenocortical adenomas. Genetic alterations of β-catenin, a Wnt pathway key component, have been described in ACT. More rarely, mutations in gsp and ACTH-receptor have been observed in ACT. The genetics of another group of adrenal diseases that can lead to adrenal nodular hyperplasia – congenital adrenal hyperplasia (CAH) and glucocorticoid-remediable aldosteronism (GRA) – have also been extensively studied. Aberrantly expressed G-protein-coupled receptors in the adrenal cortex have been reported in the ACTH independent macronodular adrenal hyperplasia (AIMAH).
Most recent achievements have unraveled the integrated genomic landscape of adrenal cortical carcinomas (ACCs) and revealed new driver genes, identified the role of mutated PKACA subunits in the pathogenesis of more than a third of sporadic adrenal cortical adenomas (ACAs) with overt Cushing’s syndrome, and discovered a new and nonanticipated gene, ARMC5 ( armadillo repeat containing 5 ), whose germline mutations cause a familial form of AIMAH.
This chapter summarizes recent advances in the genetics of ACT, highlighting both improvements in our understanding of the pathophysiology and the diagnosis of these tumors.
The particular, aldosterone-producing ACT, responsible for Conn’s adenomas, are discussed in another, specific Chapter.
Introduction
Considerable advances toward understanding the molecular mechanisms of ACT development have recently been made. The study of rare genetic syndromes associated with ACT has greatly facilitated progress and has increased our understanding of sporadic adrenal tumors. Furthermore, several observations have demonstrated that genetic alterations are frequent in both benign and malignant ACT.
The Li–Fraumeni Syndrome: TP53 and Locus 17p13
Germline mutations in tumor suppressor gene TP53 are present in 70% of families with Li–Fraumeni syndrome (LFS). This syndrome displays dominant inheritance and confers susceptibility to breast carcinoma, soft tissue sarcoma, brain tumors, osteosarcoma, leukemia, and adrenocortical carcinoma (ACC). Other possible tumors include melanoma, gonadal germ cells tumors, and carcinoma of the lung, pancreas, and prostate. These tumors have an early onset, affecting mostly children and young adults. Germline mutations in TP53 have been observed in 50–80% of children with apparently sporadic ACC in North America and Europe. The incidence of pediatric ACC is about 10 times higher in southern Brazil than in the rest of the world, and a specific germline mutation has been identified in exon 10 of the TP53 gene (R337H) in almost all cases. In sporadic ACC in adults, somatic mutations of TP53 are found in only 25% of ACC cases and are located in four “hot spot regions” within exons 5 and 8, as reported in a recent review. More recently, Libé et al., by sequencing the entire TP53 coding region, reported TP53 mutation in 33% of ACT, which is associated with a more aggressive and advanced staging. Loss of heterozygosity (LOH) at 17p13 has been consistently demonstrated in ACC but not in adrenocortical adenoma (ACA). LOH at 17p13 was recently reported to occur in 85% of ACC and only in 30% of ACA. Increased prevalence of LOH at 17p13 is correlated with higher Weiss score – an index of pathological alterations used to determine the malignancy of ACT. It has therefore been suggested that 17p13 LOH could be used as a molecular marker of malignancy in ACT: in a large prospective study of ACT patients, 17p13 LOH was demonstrated to be an independent variable predictive of recurrence after complete surgical removal of localized ACT. The discrepancy between the frequencies of TP53 mutation and 17p13 LOH may be accounted for by the existence of another tumor suppressor gene in this region. More recently, a common minimal region of loss on 17p13 in ACCs is identified, whereas no minimal region of loss in ACAs could be demonstrated. A significant downregulation of two genes that map in this region, ACADVL and ALOX15B , in ACCs compared with ACAs was demonstrated, suggesting that the ACADVL and ALOX15B expression are good discriminators between ACCs and ACAs.
The Beckwith–Wiedemann Syndrome: IGF-II (Insulin-Like Growth Factor II) and 11p15 Alterations
The 11p15 region is organized into two different clusters: a telomeric domain including the IGF-II gene, H19 , and a centromeric domain including CDKNIC ( p57kip2 ). The IGF-II gene encodes an important fetal growth factor, is maternally imprinted, and is therefore expressed only from the paternal allele. The H19 mRNA is not translated and this gene may modulate IGF-II expression. The p57kip2 gene encodes a cyclin-dependent kinase inhibitor involved in the G1/S phase of the cell cycle. The H19 and p57kip2 genes are paternally imprinted and are therefore expressed from the maternal allele only. Genetic or epigenetic changes in the imprinted 11p15 region, resulting in increases in IGF-II expression, and these alterations have been implicated in Beckwith–Wiedemann syndrome (BWS). This overgrowth disorder is characterized by macrosomia, macroglossia, organomegaly, and developmental abnormalities (in particular abdominal wall defects with exomphalos). It predisposes patients to the development of embryonal tumors – such as Wilms’ tumor – ACC, neuroblastoma, and hepatoblastoma. IGF-II mRNA is efficiently translated and malignant tumors contain large amounts of IGF-II protein, some of which is in the prohormone form.
The insulin-like growth factors system is involved in the development and maintenance of differentiated adrenocortical functions, and its role has been largely documented in ACTs. Many studies have demonstrated that IGF-II is often strongly overexpressed in malignant adrenocortical tumors, with such overexpression observed in approximately 90% of ACC. Transcriptome analysis of ACT has demonstrated that IGF-II is the gene most overexpressed in ACC by comparison with ACA or normal adrenal glands. The mechanism underlying IGF-II overexpression is paternal isodisomy (loss of the maternal allele and duplication of the paternal allele) or, less frequently, loss of imprinting with maintenance of both parental alleles but a paternal-like IGF-II gene expression pattern. The mitogenic effect of IGF-II is dependent on the IGF-I receptor, as reported by Logié et al., who demonstrated that IGF-II is involved in NCI H295R cell line proliferation and acts via the IGF-I receptor. IGF-II effects are restricted to tumors, and plasma IGF-II concentrations are usually in the normal range. In ACC, only the maternal H19 allele is expressed, so expression of this gene is abolished in most ACC displaying paternal isodisomy. Expression of p57kip2 is also abolished in ACC, but the precise role of the product encoded by this gene in the cell cycle machinery and tumorigenesis requires confirmation. Like 17p13 LOH, 11p15 LOH is associated with a higher risk of tumor recurrence, is more frequent in ACC than in ACA (78.5% vs. 9.5%), and correlates with Weiss score. These genetic abnormalities generate a mosaic-like pattern in some tumors, suggesting that the tumor is made up of different subpopulations of cells. Thus, 11p15 alterations could be used as a biological marker for predicting ACC malignancy after surgical removal of the tumor. However, 11p15 LOH seems to have a lower predictive value than 17p13 LOH.
Multiple Endocrine Neoplasia Type 1: The Menin Gene and Locus 11q13
The menin gene, located at the 11q13 locus, is thought to be a tumor suppressor gene. A heterozygous inactivating germline mutation of menin is found in about 90% of families affected by multiple endocrine neoplasia type 1 (MEN1). This is an autosomal dominant syndrome with high penetrance and an equal sex distribution. The principal clinical features include parathyroid (95%), endocrine pancreas (45%), and pituitary (45%) tumors, thymic carcinoids, and thyroid adenomas. Adrenocortical tumors and/or hyperplasia are observed in 25–40% of MEN1 patients. In most cases, they are nonfunctional ACA that can be managed conservatively with radiological/hormonal follow-up. ACC has rarely been reported in MEN1 patients.
Somatic mutation of the menin gene is very rare: as reported in a recent series, one mutation was identified in a series of 41 ACA in one study, and one mutation in a series of ACC was found in another. By contrast, LOH at 11q13 was identified in more than 90% of informative ACC, whereas it has been reported in less than 20% of informative adenomas. However, LOH in ACC involves almost the entire 11q domain, suggesting that an as yet unidentified tumor suppressor gene located on the long arm of the chromosome is involved in ACC formation. For more details, see Chapter 26 .
The Carney Complex: PRKAR1A Gene and Locus 17q22-24
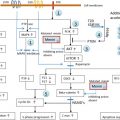
The regulatory R1A subunit of protein kinase A (PRKAR1A) is a key component of the cAMP signaling pathway that has been implicated in endocrine tumorigenesis. This gene maps to 17q22-24, a locus that has been implicated, by linkage analysis in a dominantly multiple neoplasia inherited syndrome with many clinical and pathological manifestations, the Carney complex (CNC). Heterozygous inactivating germline mutations of PRKAR1A have been demonstrated in about 45–65 % of CNC families. LOH at 17q22-24 is observed in tumors from CNC patients, suggesting that PRKAR1A is a tumor suppressor gene. The main features of CNC include spotty skin pigmentation (lentiginosis), endocrine overactivity, and cardiac myxomas. The tumors observed in CNC patients include growth-hormone (GH)-secreting pituitary adenoma, thyroid adenomas or carcinomas, testicular tumors (large-cell calcifying Sertoli cell tumors), ovarian cysts, melanocytic schwannomas, breast ductal adenomas, and adrenocortical lesions. ACTH-independent Cushing’s syndrome caused by primary pigmented nodular adrenocortical disease (PPNAD) is observed in 25–30% of patients with CNC. PPNAD is caused by a primary bilateral adrenal defect and may occur in patients with no other CNC features and no family history of CNC. ACTH-independent Cushing’s syndrome is often atypical in PPNAD: it may be cyclic, associated with a paradoxical increase in cortisol concentration after dexamethasone administration, and may be found in patients with normal CT scans. The frequency of PRKAR1A mutations is about 80% in CNC patients with Cushing’s syndrome, suggesting 17q22-24 defects are more likely to be found in families with PPNAD. Moreover, patients with isolated PPNAD and no family history of CNC may present de novo germline mutation of PRKAR1A . Somatic mutation of PRKAR1A can occur in PPNAD, inactivating the wild-type allele in a patient already presenting a germline mutation.
Somatic PRKAR1A mutations have been also demonstrated in sporadic secreting ACA, with clinical, hormonal, and pathological characteristics similar to those of PPNAD. LOH at 17q22-24 has been also observed in sporadic ACA and seems to be restricted to the PRKAR1A locus, suggesting the possible involvement of this tumor suppressor gene. By contrast, LOH seems to affect a large part of 17q in ACC, suggesting that PRKAR1A alteration may play only a minor role in malignant ACT growth.
Recently, a whole-genome association study led to the discovery of a new actor of the cAMP pathway involved in adrenal cortex tumorigenesis, the phosphodiesterase 11A4 ( PDE11A ) gene. PDE11A inactivating mutations have been reported in a subgroup of patients with Cushing’s syndrome due to micronodular adrenal hyperplasia (MAH). Moreover, two functional missense substitutions were identified, suggesting a role of PDE11A as genetic predisposition factor. These results has been confirmed by Libé et al. in a large cohort of ACT (ACA, ACC, and AIMAH): in fact, a higher frequency of missense variants were found in particular in the AIMAH group and a higher frequency of synonymous polymorphisms were found in ACC.
Familial Adenomatous Polyposis (FAP) and the β-Catenin Gene; and the Lynch Syndrome
Genetic alterations of the Wnt signaling pathway were initially identified in familial adenomatous polyposis coli (APC) and have been extended to a variety of cancers. ACT have been observed in some case reports of patients with familial APC. Furthermore, familial APC patients with germline mutations of the APC gene that lead to an activation of the Wnt signaling pathway may develop ACTs. Molecular studies have suggested that somatic mutations of APC could occur in these tumors in patients already having a germline defect.
The Wnt signaling pathway is normally activated during embryonic development. β-Catenin is a key component of this signaling pathway. It has a structural role in cell–cell adhesion, and is a transcription cofactor with T-cell factor/lymphoid enhancer factor (TCF/LEF) mediating transcriptional activation of target genes of the Wnt signaling pathway.
Interestingly, gene profiling studies in various types of ACTs have shown the frequent activation of Wnt signaling target genes: in ACC a microarray approach has shown that ectodermal-neural cortex one (ECN-1) was upregulated. In both benign and malignant ACT, β-catenin accumulation can be observed. These alterations seem very frequent in ACC, consistent with an abnormal activation of the Wnt-signaling pathway. This is explained in a subset of ACT by somatic mutations of the β-catenin gene altering the glycogen synthase kinase 3-β (GSK3-β) phosphorylation site. GSK3-β is implicated in the regulation of β-catenin. In the absence of Wnt signaling, the level of β-catenin is low: β-catenin is phosphorylated at critical NH2-terminal residues by the GSK3-β bound to a scaffolding complex of axin and adenomatous polyposis proteins (APC), and subsequently the phosphorylated protein is degraded by the ubiquitin–proteasome system. Wnt stimulation leads to the inactivation of GSK3-β and thereby the stabilization of β-catenin in the cytoplasm. In the Lynch syndrome, DNA instability is associated with an array of tumors, primarily in the gut. Yet evidence has been provided that rare ACTs may also be part of the syndrome.
The McCune–Albright Syndrome: GNAS1 Gene
The trimeric G protein (α, β, γ subunit) is responsible for transmembrane signal transduction following ligand activation of a G-protein-coupled seven-transmembrane domain adrenocorticotropin receptor (ACTH-R). Somatic activating mutations of the GNAS1 gene (mutant Gs protein, termed gsp ) responsible for excess activity of the cyclic AMP (cAMP) signaling pathway have been reported in McCune–Albright syndrome (MAS). This disease is characterized by polyostotic fibrous dysplasia, café au lait spots, precocious puberty, and hyperfunction of multiple endocrine glands (thyroid, adrenal glands, and pituitary gland). Hypercortisolism occurs in 5% of patients and is due to ACTH-independent macronodular adrenocortical hyperplasia (AIMAH). In MAS, the gsp mutation occurs during embryonic development, as demonstrated by its mosaic pattern of distribution in various tissues. Few somatic GNAS mutations have been found in ACTs, as reported in a recent review.
Two different gsp mutations have been reported in three patients with Cushing’s syndrome due to AIMAH without MAS features. The authors speculated as to whether these patients presented a disease in the spectrum of MAS, with a late somatic mutation leading to a single defect, or whether they were the first reported cases of isolated AIMAH with gsp mutations involved in molecular pathogenesis.
Congenital Adrenal Hyperplasia
CAH is one of the most frequent genetic endocrine diseases, inherited as an autosomal recessive trait. It is caused by the loss of or severe decrease in activity of one of the steroidogenic enzymes involved in cortisol biosynthesis (mostly 21-hydroxylase (21-OH), 11β-hydroxylase (11β-OH), and 3β-hydroxysteroid dehydrogenase). Deficiencies in 21-hydroxylase ( CYP21 ) are the most common causes of CAH, accounting for 90–95% of cases. All the known biochemical defects impair cortisol secretion, resulting in the stimulation of pituitary corticotrophs, leading to compensatory hypersecretion of ACTH, resulting in hyperplasia of the adrenal cortex. In the past, both homozygous and heterozygous patients with CAH have been reported to have substantially enlarged adrenal glands and a prevalence of adrenal incidentalomas. No mutation in CYP21 was detected in a study of leukocyte DNA from a series of 27 patients, whereas two heterozygous CYP21 mutations were found in adrenal tumor DNA. By contrast, in another series, a higher frequency of classic CAH carriers (16%) and of manifest CAH (2%) was reported among patients with adrenal adenomas than in the general population. For more details, see Chapter 18 .
Glucocorticoid-Remediable Aldosteronism
GRA was the first described familial form of hyperaldosteronism. This disorder is characterized by the chronic regulation of aldosterone secretory function by ACTH. Aldosterone hypersecretion can therefore be chronically blocked by exogenous glucocorticoids, such as dexamethasone. This autosomal dominant disorder has been shown to be caused by a hybrid gene formed by crossover between the ACTH-responsive regulatory portion of the 11β-hydroxylase ( CYP11B1 ) gene and the coding region of the aldosterone synthase gene ( CYP11B2 ). Adrenal tumors, together with micronodular and homogeneous hyperplasia of the adrenal cortex, have been observed in the familial cases.
ACTH Receptor ( ACTH-R ) Gene
ACTH-R belongs to a subgroup of five receptors of the G-protein-coupled receptors superfamily. This subgroup consists of ACTH-R (or MCR-2), MSH-R (MCR-1), and three other receptors (MCR3 to 5). It is encoded by an intron-less gene on chromosome 18p11.2. Inactivating mutations in ACTH-R have been identified in several families with hereditary isolated glucocorticoid deficiency. Screening for ACTH-R mutations in a variety of adrenal tumors has identified no somatic activating mutations to date. Swords et al. reported the functional characterization of a potential activating germline mutation of ACTH-R and demonstrated that it displays high levels of basal activity due to a defect in receptor desensitization. ACTH-R LOH has also been investigated in ACA and ACT: it was observed in two of four informative cancers, but not in 15 hyperfunctioning adenomas, suggesting a role for ACTH-R in cellular differentiation.
ACTH-Independent Macronodular Adrenal Hyperplasia (AIMAH)
ACTH-Independent macronodular adrenal hyperplasia (AIMAH) is a rare cause of endogenous Cushing’s syndrome, in which clinical features usually become apparent only after several decades of life. The pathophysiology of this entity is heterogeneous and has been intensely explored in recent years. Several G-protein coupled receptors aberrantly expressed in the adrenal cortex have been implicated in the regulation of steroidogenesis and in the initial cell proliferation in AIMAH, such as the gastric inhibitory polypeptide receptor (GIP-R), β-adrenergic receptors, vasopressin (V2–V3) receptors, serotonin (5-HT7) receptor, and probably angiotensin II receptor (AT1R). Increased expression or over activity of eutopic receptors such as those for vasopressin (V1) receptor, luteinizing hormone/human chorionic gonadotropin (LH/hCG-R), serotonin (5-HT4) receptor, and leptin receptor.
The molecular mechanisms leading to the abnormal expression of eutopic and ectopic receptors in the adrenal glands of patients with AIMAH and, less commonly in ACAs, remains incompletely understood. The GIP-R is the more extensively characterized ectopic receptor in the adrenal CS. It was unclear whether aberrant hormone receptors are a primary phenomenon responsible for the pathogenesis of AIMAH or adenomas, or an epiphenomenon resulting from cell proliferation and dedifferentiation; there are now several evidences in favor of the former hypothesis.
Initially reported cases of AIMAH appeared to be sporadic; more recently, first-degree relatives, screenings identified several familial cases with an autosomal dominant pattern of transmission. Up to now, the genetic basis of these familial forms has not been extensively studied. Moreover, the potential presence of aberrant receptors was evaluated only in the recently studied families. As reported in a recent review, some aberrant receptors have been identified so far: V1-vasopressin and β-adrenergic in one family; β-adrenergic in a second one; V1–V2 and V3-vasopressin in another family, and combined 5HT4 and V1–V2-vasopressin in another one. A systematic clinical screening of a family with hereditary cortisol-secreting β-adrenergic responsive AIMAH revealed unsuspected subclinical Cushing’s syndrome and aberrant β-adrenergic regulation of cortisol in all familial cases studied with subclinical Cushing’s syndrome.
In a most recent paper describing the very thoughtful approach of Hervé Lefebvre’s team, the pathophysiological role of ACTH locally produced by the tumoral adrenocortical cells themselves has been convincingly demonstrated. This intracrine action of ACTH should logically modify the name of these tumors, which are indeed “ACTH- dependent” if not “pituitary-dependent,” Primary macronodular adrenocortical hyperplasian (PMAH) seems therefore more appropriate.
Assié G. et al. have established that germline mutations of ARMC5 ( armadillo repeat containing 5 ) was responsible for a familial form of benign primary macronodular adrenal hyperplasia with Cushing’s syndrome. This unanticipated new actor is a tumor suppressor gene, the normal allele being functionally lost, by different ways, in each of the many adrenal nodules in the same patient. Mutations of ARMC5 have been confirmed by other groups. Familial screening now allows early diagnosis of mutated carriers who can be closely and prospectively followed clinically. It remains to be established whether mutated ARMC5 also acts, in some way, through the pivotal same cAMP pathway.
Sporadic Adrenocortical Adenomas With Cushing’s Syndrome
Beuschlein’s group published the molecular mechanism responsible for more than a third of sporadic adrenocortical adenomas with clinically overt Cushing’s syndrome: somatic, activating mutations of the gene coding for the PKA catalytic subunit (PKACα). This mutation was primarily found by exome sequencing in eight of 10 such tumors, and subsequently screened in more than 200 adrenal cortical tumors of various types, within the ENS@T (European Network for the Study of @drenal Tumors) collection of stored tumor specimens. Remarkably one single hot spot mutation, the c.617A>C single nucleotide variation, inducing the p.Leu206Arg substitution, was reported by far as the most common alteration in the cortisol-secreting adenomas (95%). There are many lessons from this discovery: the PKACα mutations are strictly related to adrenocortical adenomas with overt Cushing’s syndrome; they are not found in ACC, nor in aldosterone producing adenomas; quite unexpectedly they are not found either in patients with subclinical hypercortisolism, highly suggesting that these rather frequent adenomas, often diagnosed as “incidentalomas,” are a different pathophysiological entity and not merely an early form of overt hypercortisolism; there remain other molecular causes to be found, in the two-thirds of “nonmutated” tumors with overt Cushing’s syndrome, and in all the others; it is remarkable that few mutations were present in adenomas (an average of five by tumor), quite different from the high numbers in ACCs (see further): these benign, homogeneous, tumors are ideal biological specimens for genetic discoveries. In the same paper, the authors report on five patients with cortisol-secreting bilateral adrenocortical hyperplasia who had copy number gains on the PKACα in their constitutive DNA. In vitro studies demonstrated the allelic dosage effect of PKACα as an alternative mechanism to activate the cAMP pathway. Indeed, these data further emphasize the pivotal role of this pathway, which, through different and exclusive actors, can provoke a variety of benign – most often with cortisol hypersecretion – ACT.
Beuschlein’s paper was “rapidly” – within weeks! – confirmed by three other independent teams, in high level journals.
Integrated Genomic Organization of ACCs
Assié et al. reported the first “integrated genomic organization of adrenocortical carcinoma.” This exhaustive work was accomplished by the combination of a well-organized and rather exceptional collection of these rare tumors, more than 122 specimens collected within ENS@T, and the use of the most actual genomic approaches: exome sequencing, genome, miRnome, transcriptome, and methylome analyses. These long-time and European-wide efforts ultimately allowed drawing the “genomic landscape” of these rare but devastating endocrine tumors. ACCs have a high rate of mutations (median of 24 per tumor, excluding two tumors accounting together for 1881 mutations!); interestingly, these mutations point to few definite pathways: the Wnt/βcat pathway activation is the most frequently involved, and, among its actors, a new tumor suppressor gene was identified, ZNRF3 , which is altered in up to 21% of the tumors. Along the same pathway, CTNNB1 is also frequently mutated (16%), and the ZNRF3 / CTNNB1 alterations are mutually exclusive, stressing both the high prevalence and the pathophysiological consequences of altered Wnt/βcat pathway in ACCs; gene alterations in other known driver genes were confirmed ( TP53 , CDKN2A , RB1 , and MEN1 ), and new ones unraveled ( DAXX , TERT , and MED12 ), each being found in more than 5% of ACCs. Altogether these recurrent gene alterations were present in ca. 60% of the tumors. The integrated genomic approach also studied the miRNA profiles and the methylation patterns of the tumors. Remarkably, it separated different groups of tumors, with different prognoses, which also correlated nicely with previous transcriptomic data obtained 5 years earlier in the same tumors. These data convincingly show that ACCs can now be classified on a molecular basis, which impact on the prognosis and ultimately on personalized treatment.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


