GENETICS
The autosomal recessive mode of inheritance of adrenal steroidogenic defects was recognized in the early 1950s.62,63 and 64 The linkage of 21-hydroxylase deficiency to the HLA complex on chromosome arm 6p was discovered in the late 1970s,65 and by the mid-1980s, further molecular genetic details had been unravelled.66 Other human adrenal steroidogenic defects also have been successfully subjected to molecular genetic analysis.
21-HYDROXYLASE DEFICIENCY
The CYP21 (CYP21B) structural gene encoding steroid 21-hydroxylase and a pseudogene (CYP21P or CYP21A) are located 30 kilobases (kb) apart in the HLA complex on chromosome band 6p21.3.67,68 Although the two genes are 98% identical in nucleotide sequence, CYP21P has accumulated a number of mutations that render any gene product completely inactive. These include an 8-base-pair (bp) deletion in exon 3, a frame shift in exon 7, and a nonsense mutation in exon 8.
Most mutations causing 21-hydroxylase deficiency are caused by apparent recombinations between CYP21 and CYP21P. Approximately 20% of these are unequal meiotic crossovers resulting in a deletion of a 30-kb DNA segment that includes the 3′ end of the pseudogene and the greater portion of the active gene.69 This deleted haplotype is incapable of producing any active enzyme. About 80% of mutations result from apparent gene conversions that transfer small segments containing one or more deleterious mutations from CYP21P to CYP21. The most common of these, accounting for an additional ˜25% of mutant haplotypes, is a single A→G transition at nucleotide 656 that causes abnormal pre–messenger RNA (mRNA) splicing. Less commonly found are missense mutations, which cause changes in the protein’s amino-acid sequence.
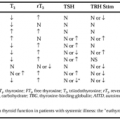
Several large studies have examined the prevalence of individual mutations in an attempt to correlate specific mutations with particular clinical manifestations of the disease.70,71,72,73 and 74 These correlations are most reliably made in individuals who are homozygous or hemizygous (the other chromosome carries a deletion) for each mutation. Table 77-3 illustrates the mutations commonly found in classic and nonclassic forms of 21-hydroxylase deficiency, grouped into three categories according to the predicted level of enzymatic activity based on in vitro mutagenesis and expression.75,76,77 and 78 Group A, with total ablation of enzyme activity, is most often associated with salt-wasting disease; group B, with 2% normal activity, consists predominantly of patients with simple virilizing disease; and group C, with 20% to 60% of normal activity, is most often associated with the nonclassic disorder.
These studies suggest that mutant CYP21 enzymes carrying discrete amino-acid substitutions identified in CAH patients exhibit in vivo activities that are generally consistent with in vitro predictions, and correlate with disease severity. Exceptions to this rule include patients with moderate or severe mutations and relatively mild disease. Because aldosterone is normally secreted at a rate 100 to 1000 times lower than that of cortisol, residual enzyme activity as low as 0.6% of normal activity, as seen in the Ile-172→Asn nonconservative substitution, allows enough aldosterone synthesis to prevent symptoms of salt wasting, resulting in the simple virilizing phenotype. Factors outside the CYP21 locus may also influence development of the salt-wasting phenotype.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


