Abstract
Genetic skeletal disorders make up one of the largest groups among rare inherited conditions. Referred to as skeletal dysplasias, these hereditary diseases of bone may be classified according to radiologic anatomic appearance, density of bone, and state of mineralization. This chapter examines a number of selected skeletal dysplasias representing each of the three groups: (1) sclerosing bone disorders defined as thickening either of trabecular or cortical bone; (2) disorders of defective mineralization with low bone mass called either rickets in childhood or osteomalacia in adults; and (3) dysplasias of bone and cartilage with normal or low bone mass, called osteopenia or osteoporosis in the absence of a defect in mineralization. In the absence of unique radiographic changes, diseases can be classified using a combination of bone histology, serum chemistries, and genetic mutations.
Keywords
skeleton, dysplasia, sclerosis, osteomalacia, rickets, genetic, mutation, fracture, osteopetrosis, mineralization
Introduction
Genetic skeletal disorders make up one of the largest groups among rare inherited conditions. Referred to as skeletal dysplasias, these hereditary diseases of bone may be classified according to radiologic anatomic appearance, density of bone, and state of mineralization. This chapter examines a number of selected inherited skeletal dysplasias representing each of the three groups: (1) sclerosing bone disorders defined as thickening either of trabecular or cortical bone; (2) disorders of defective mineralization with low bone mass called either rickets in childhood or osteomalacia in adults; and (3) dysplasias of bone and cartilage with normal or low bone mass, called osteopenia or osteoporosis in the absence of a defect in mineralization. In the absence of unique radiographic changes, diseases can be classified by bone histology, serum chemistries, or genetic mutations. This chapter discusses representative disorders ( Table 12.1 ) from each of the three major categories of genetic sclerosing bone disorders, mineralization defects, and other dysplasias of bone and cartilage with normal or low bone mass.
| Sclerosing bone disorders Inherited sclerosing dysplasias
|
Bone dysplasias with defective mineralization
|
Selected inherited dysplasias of bone and cartilage
|
Although cases of most rare bone disorders have been studied in detail, effective therapeutic interventions are lacking for many of these conditions. Given that most of the diseases begin in infancy or childhood and are irreversible, the patients are given the dim prospect of lifelong suffering. It is hoped that the advances in basic science of the past two decades can be used in innovative ways to develop much-needed effective therapeutics.
Sclerosing bone disorders
Sclerosing bone dysplasias vary in severity and present with a wide range of radiologic, clinical, and genetic features. Hereditary sclerosing bone dysplasias result from disturbance(s) in pathways involved in osteoblast or osteoclast function, leading to abnormal accumulation of trabecular bone, called osteosclerosis, or increased cortical bone, called hyperostosis. Investigations into the genetic basis of inherited sclerosing bone dysplasias have resulted in identification of gene mutations that may alter critical skeletal developmental or bone cell regulatory functions through activating or inactivating mutations.
Knowledge of the radiologic appearances, distribution, and associated clinical findings with increased bone density are crucial for accurate diagnosis. Acquired disorders with increased bone density may simulate genetic sclerosing bone dysplasias, including hepatitis C-associated osteosclerosis, osteoblastic metastases, Paget’s disease of bone, myelofibrosis and sickle cell disease. Selected inherited disorders are discussed in the subsequent section.
Osteopetrosis
Osteopetrosis is a group of closely related inherited disorders due to the loss of osteoclastic bone resorption from either the absence of osteoclasts or functional loss. The central features include fractures, dense bone, low to absent modeling, obliteration of the marrow space, and anemia with extramedullary hematopoiesis. The condition is also known as marble bone disease from the radiologic appearance of the solid, dense skeleton, which was initially described by Albers-Schonberg in 1904. Current terminology uses Albers-Schonberg disease to refer to the more benign adult onset form of osteopetrosis, also called autosomal dominant osteopetrosis type II. The same phenotype is also produced by carbonic anhydrase (CA) II deficiency, which is transmitted by an autosomal recessive pattern of inheritance characterized by a milder course than the severe form of osteopetrosis and accompanied by renal tubular acidosis, mental retardation, and cerebral calcification.
The distinctive forms of osteopetrosis differ by the mode of inheritance, age of appearance, and severity of the disease. The autosomal dominant forms are generally more benign with adult onset and minimal disability or symptoms. The autosomal recessive type appears in infancy and follows a malignant course with high mortality rate. There is a neuropathic subtype associated with mutations in OSTM1 or CLCN7 leading to seizures, developmental delay, hypotonia, and renal atrophy. A rare autosomal recessive form with onset in childhood is intermediate in severity with a more benign course.
Clinical Presentation
The incidence of the severe autosomal recessive malignant osteopetrosis ranges from one in 200,000 to one in 500,000 live births. Hypocalcemia in infants and children is the result of absent osteoclastic bone resorption with loss of bone modeling. Failure of bone modeling may also cause neurologic abnormalities of the cranial nerves, including paralysis due to narrowing of cranial foramina.
Adult benign autosomal recessive osteopetrosis (ARO) is often diagnosed by the presence of typical skeletal changes seen in radiographs taken to evaluate fractures. The prevalence is one in 100,000 to one in 500,000 adults. The course may not be benign, with fractures accompanied by loss of vision, deafness, mandibular osteomyelitis, and other complications often more characteristic of the juvenile or intermediate form of the disease. The pattern of inheritance is not always predictable with examples of nonpenetrance resulting in skipped generations, or in contrast, severely affected children being born into families with a largely benign form of the disease.
Radiography
The major radiographic change in osteopetrosis is a systemic increase in bone mass with thickening of both trabecular (sclerosis) and cortical bone (hyperostosis) ( Fig. 12.1 ). Metaphyseal areas of bone may contain alternating dense (sclerotic) and radiolucent banding. Metaphyses may widen and create an “Erlenmeyer flask” deformity. Pathologic fractures may be discovered at any site in the skeleton. Changes in the growth plates associated with rickets may be present due to secondary hyperparathyroidism prompted by hypocalcemia. The basal aspects of the skull is thickened, and there is thickening of other skull bone with narrowing of the cranial nerve foramina and minimal or absent development of the mastoid and paranasal sinuses. Nuclear medicine Tc-99m scans reveal fracture sites and complications such as minimal bone marrow space and osteomyelitis.
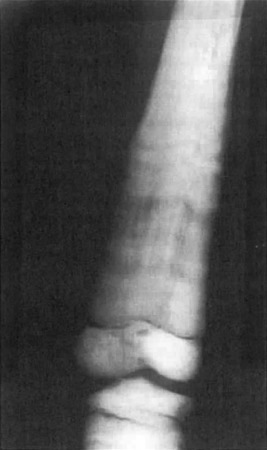
Laboratory Tests
Serum calcium may be decreased in severe disease and accompanied by increased serum parathyroid hormone and 1,25-dihydroxyvitamin D levels. Serum levels of osteoclastic tartrate-resistant acid phosphatase (TRAP) and the brain isoenzyme creatine kinase may be increased.
Histology
The presence of small areas or “islands” of persistent calcified cartilage within trabecular bone is a unique marker for near absence of osteoclast function during endochondral bone formation. The heterogeneity of osteopetrosis is indicated by the osteoclast number, which may be increased or normal. Rarely is the number of osteoclasts decreased. When nuclear number is increased, the ruffled border and clear zone will be absent. The bone matrix may have multiple areas of immature woven bone.
Genetic Pathophysiology
About 50% of patients with autosomal recessive malignant infantile osteopetrosis have a mutation of the TCIRG1 gene, which encodes for the osteoclast-specific α3 subunit of the vacuolar proton pump. The pump is responsible for acidification of the microspace of the bone mineral surface enclosed by the osteoclast ruffled border. Autosomal dominant osteopetrosis type II (Albers-Schonberg disease) results from a mutation in the CLCN7 chloride channel gene that disables osteoclast resorptive function. CLCN7 mutations can also cause autosomal recessive, malignant, or the intermediate osteopetrosis. Mutations of the CAII gene cause carbonic anhydrase II deficiency and dysregulation of acidification, normal function of which is required for normal osteoclast resorption. Rarely, infants with osteopetrosis have an inactivating mutation in the receptor activator of nuclear factor- k B ligand (RANKL) gene. Such patients have low osteoclast counts and fail bone marrow transplant therapy.
Genetic Diagnostic Testing
Mutation analysis can be used to identify severe cases caused by mutations of the TCIRG1 and CLCN7 genes. The genetic basis of the disease is now largely uncovered by identification of mutations in TCIRG1 , CLCN7 , OSTM1 , SNX10, CSF-I , and PLEKHM1 , which lead to osteoclast-rich ARO in which osteoclasts are abundant but have severely impaired resorptive function. In contrast, mutations in TNFSF11 and TNFRSF11A lead to osteoclast-poor ARO. In osteoclast-rich ARO, impaired endosomal and lysosomal vesicle trafficking result in defective osteoclast ruffled-border formation and, hence, the inability to resorb bone and mineralized cartilage.
Treatment
Allogeneic human leukocyte antigen-identical bone marrow transplantation has the highest success rate. Other children may respond to interferon-gamma-1β, 1,25-dihydroxyvitamin D3, methylprednisolone, and a low calcium-high phosphorus diet. Bisphosphonate therapy may increase bone density and decrease fracture rates, but does not correct the underlying genetic defects. Surgical treatment is indicated to correct skeletal deformities and stabilize fractures to optimize healing.
Progressive Diaphyseal Dysplasia
The disorder also known as Camurati–Engelmann disease is an autosomal dominant inherited disease notable for symmetrical thickening and widening of the endosteal and periosteal surfaces of the diaphysis of long bones, primarily the femur and tibia and less frequently the fibula, radius, and ulna.
Clinical
The disorder presents during childhood with lower extremity pain and tenderness of the involved areas, fatigue, muscle wasting, and weakness, often manifest as a waddling gait. On examination, the limbs are thin due to loss of muscle mass while the bones are prominent and tender. Skull involvement may also be present with a large cranium, prominent forehead, proptosis, cranial nerve palsies, and hydrocephalus with increased intracranial pressure. There may be delayed puberty with central hypogonadism. While skeletal radiographic studies show progressive bone changes with time, the clinical manifestations may be quite variable with occasional spontaneous improvement, more likely during the adult years. Some patients with more severe disease may have Raynaud’s phenomenon, hepatosplenomegaly, and other changes suggestive of vasculitis.
Radiography
The hyperostosis is progressive and seen as a patchy, irregular thickening of both the endosteal and periosteal surfaces due to new bone formation ( Fig. 12.2 ). The areas spread symmetrically along the diaphysis of long bones with eventual involvement of the metaphysis. Tc-99m bone scan radiotracer uptake is increased in radiographically involved areas. The presence of marked tracer uptake at sites with minimal radiographic changes suggests early skeletal disease.
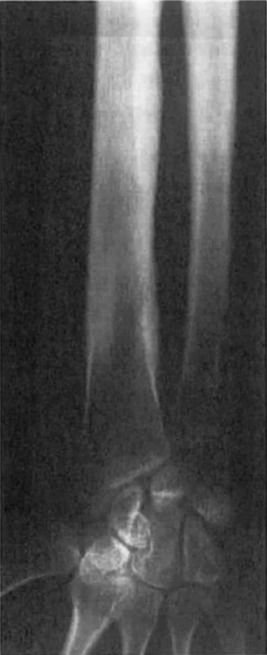
Laboratory
Metabolic studies show markedly positive calcium balance, which is manifest as a tendency for a mild hypocalcemia and marked hypocalciuria, consistent with increased renal tubule calcium reabsorption and positive calcium balance. Some patients have elevation of serum alkaline phosphatase and urine hydroxyproline excretion, indicating increased bone turnover. Elevated erythrocyte sedimentation rate, leukopenia, and mild anemia in some patients may indicate that the clinical and laboratory changes of inflammation are due to an accompanying systemic connective tissue inflammatory disorder.
Histology
Histologic examination of bone shows new bone formation along the diaphyseal surfaces. Woven bone can be seen undergoing maturation and incorporation into cortical bone. Affected muscle shows both myopathic and vascular changes.
Genetics and Pathophysiology
The genetic defect is caused by various activating mutations at a specific location (chromosome 19q13.2) within one region of the gene that encodes for transforming growth factor (TGF)-β1. The most common mutation reported is an arginine-cysteine amino acid change at codon 218 (R218C) in the latency-associated peptide domain of TGF-β1. In one study in vitro peripheral blood mononuclear cells obtained from three related CED patients harboring the R218C mutation, TGF-β1 bioactivity, and osteoclast formation in vitro were increased and inhibited by addition of the soluble TGF-β1 type II receptor. Increased osteoclast activity is consistent with clinical and biochemical evidence of increased resorption. In addition, the mutation in bone cells activates TGF-β1 resulting in overstimulating of bone matrix formation. Binding of a mutated latency-associated peptide domain promotes the persistence of the TGF-β1 action.
Genetic Diagnostic Testing and Interpretation
Characteristic radiologic changes strongly support the diagnosis, which can be confirmed by the identification of an activating mutation within the TGF-β1 gene.
Treatment
Small doses of glucocorticoids (prednisone five mg daily) may relieve bone pain and reverse the abnormal histologic changes of bone formation. There is limited experience with bisphosphonates, and no random controlled trials have been done. However, clinical improvement has been reported in a small number of patients. Treatment with the angiotensin II type 1 receptor antagonist Losartan relieved bone pain, increased activity, and increased body composition with increased lean and fat mass during 38 months of therapy in a nine-year-old girl.
Pyknodysostosis
Pyknodysostosis is a rare autosomal recessive disorder caused by an inactivating mutation in the lysosomal cathepsin K ( CTSK ) gene and characterized clinically by osteosclerosis, fragility fractures, short stature, dysmorphic facial features, dental abnormalities, distal phalangeal acroosteolysis, and delayed closure of cranial sutures. There is a range of age at presentation from childhood to later adult years contributing to an underdiagnosis or misdiagnosis of this form of short-limb dwarfism. The condition is thought to be a form of osteosclerosis as both have progressive generalized osteosclerosis and recurrent fractures. Pyknodysostosis may have been responsible for the short stature and other physical features of the French impressionist painter Henri de Toulouse-Lautrec.
Clinical
Features include short stature, kyphoscoliosis, chest deformity, high arched palate, blue sclera, and proptosis. Dysmorphic facial features include small face and chin, pointed nose, large cranium with fronto-occipital prominence, persistence of the open anterior fontanel, and a large angle of the mandible. Extremities are notable for short upper and lower extremities, small square hands, and hypoplastic nails.
Radiography
Radiographs reveal a generalized increase in bone density with normally shaped long bones, in contrast to the appearance of the long bones in osteopetrosis. Osteosclerosis appears in childhood and increases with age. There are recurrent fractures. Cranial sutures are separated with persistent patency of the anterior fontanel. Hypoplasia of the sinuses, mandible, distal clavicles, ribs, and terminal phalanges are variable findings. Other common cranial features include persistence of deciduous teeth and sclerosis of the calvarium and base of the skull. Long bones have narrow medullary canals.
Laboratory
Bone-related chemistries include normal serum calcium, phosphorus, and alkaline phosphatase. Unlike osteopetrosis, there is no anemia. In a report of six affected children, growth hormone and serum insulin-like growth factor 1 levels were low in five.
Histology
There is normal cortical bone architecture with sclerosis of trabecular bone. Osteoblastic bone formation and osteoclastic bone resorption activities are decreased. Osteoclasts are present in the sclerotic trabecular bone but do not function normally.
Genetic Pathophysiology
The molecular basis for the disorder is a mutation in the CTSK gene that encodes cathepsin K, a lysosomal metalloproteinase that is highly expressed in osteoclasts. By 2010, some 27 different mutations have been reported in members of 34 unrelated families. The major defect in the disorder appears to be an impaired degradation of matrix collagen, as the cathepsin K enzyme plays an important role in digestion of matrix proteins during osteoclastic bone resorption. Electron microscopy of bone suggests that degradation of bone collagen may be defective, related to the inactive cathepsin K enzymatic digestion. Inclusion bodies have been described in chondrocytes, and virus-like inclusions have been reported in osteoclasts, but their significance remains unknown.
Diagnosis
The diagnosis is usually made during infancy or early childhood based on the major dysmorphic features of short stature, large cranium, and dysplastic facies. Suggestive clinical characteristics can be confirmed by radiographic examination of the entire skeleton and skull. As some features of osteopetrosis may be present, the differential diagnosis includes osteopetrosis, osteoporosis, cleidocranial dysplasia, and idiopathic or localized acroosteolysis of the distal phalanges.
Treatment
There is no known treatment for this disorder, and there are no reports of bone marrow transplantation. Five to 12 years of growth hormone therapy in three children with pyknodysostosis improved longitudinal growth that approached predicted levels and restored body proportion. Management is symptomatic and multidisciplinary, including orthopedic treatment of fractures and maxofacial reconstruction. Long bone fractures tend to be transverse and heal satisfactorily. But delayed union of the long bones may be complicated by large callus formation. The skeletal hardness from the underlying disease makes dental extractions and internal fixation of the long bones difficult. Vertebral surveillance is indicated to detect frequent spondylolisthesis. The prognosis is favorable, and the disease is not progressive.
Hyperostosis Corticalis Generalisata
This autosomal recessive disorder, also known as van Buchem disease, is characterized by endosteal hyperostosis in which osteosclerosis is found in the skull, ribs, and clavicles. A more severe form of endosteal hyperostosis is accompanied by syndactyly and tallness and is known as sclerosteosis. The latter affects predominantly those of Dutch ancestry, including Afrikaners. An autosomal dominant variant of van Buchem disease is called Worth disease, which is a mild form of endosteal hyperostosis.
Clinical
Narrowed cranial foramina are responsible for the major clinical manifestations including optic atrophy, deafness, and facial paralysis. These symptoms may be discovered as early as infancy in the severe form. In the milder form of the disorder, an enlarged mandible may be the initial manifestation and develops during puberty or later in some adults. Long bones may be painful, but fractures are uncommon. In contrast, syndactyly only appears in the more severe sclerosteosis. Sclerosteosis patients are tall and heavy and with time develop an enlarged cranium with facial palsy and disfigurement. Deafness may also develop. They may eventually develop increased intracranial pressure with headache and brainstem compression. Syndactyly may develop either by cutaneous growth or bony fusion. Middle and index fingers are most commonly affected.
Histology
There is uncoupled bone turnover with increased osteoblastic bone formation and low osteoclastic bone resorption leading to an accumulation of normal bone. The initial report by van Buchem described bone of normal quality.
Bone histomorphometry in one patient with sclerosteosis showed an affected skull with increased bone formation, thickened trabeculae, and accumulation of osteoid. In contrast, osteoclastic bone resorption was reduced.
Radiography
Endosteal thickening creates dense diaphyseal cortical bone with narrow medullary space. Modeling of the long bones is normal.
Osteosclerosis causes mandibular enlargement with density of the base of the skull, facial bones, vertebral bodies, pelvic bones, and ribs ( Fig. 12.3 ). In sclerosteosis, skeletal radiographs appear normal in early childhood except for the syndactyly. With progressive cortical bone growth, there is widening of the mandible and skull and greater density of the vertebral pedicles, ribs, pelvis, and long bones.
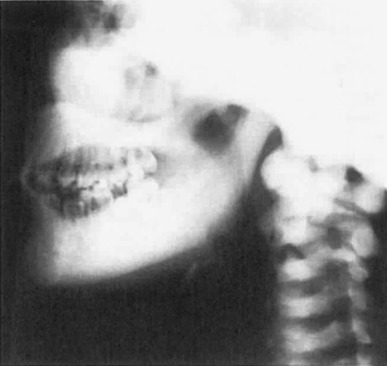
Laboratory
In both disorders, serum bone-specific alkaline phosphatase levels may be elevated, reflecting the high bone formation. Serum calcium and phosphorus are normal. Serum sclerostin levels are reduced in both van Buchem disease and sclerosteosis. The lower levels in sclerosteosis may account for the more severe disease with greater osteoblastic activity.
Genetic Pathophysiology
Sclerostin is a 213 amino acid product of SOST whose physiologic function is to bind LRP5 and LRP6 and BMP2 and other BMPs and antagonize Wnt signaling. As a result, sclerostin inhibits bone formation. Both van Buchem disease and sclerosteosis are autosomal recessive genetic diseases in which defects in the SOST gene have been assigned to the same region of the chromosome 17q12-21. However, recent advances show that the two diseases have different genetic defects. Sclerosteosis is caused by a loss-of-function mutation of the SOST gene, with increased osteoblastic bone formation and minimal osteoclastic bone resorption. Calcium homeostasis and pituitary function are normal. Van Buchem disease results from a homozygous deletion of a 52-kb regulatory element 35 kb downstream of the SOST gene that diminishes the downstream magnification of SOST .
Genetic Diagnostic Testing
The specific diagnosis of hyperostosis corticalis generalisata and sclerosteosis can be obtained by SOST gene sequencing, which is available through a select number of academic medical centers and commercial laboratories. The procedures include sequencing the SOST gene and analyses for duplication or deletion of regions of SOST .
Treatment
There is no specific medical therapy that can change the progression of the endosteal hyperostosis. Losartan, an angiotensin II type 1 receptor antagonist, has been found to downregulate the expression of TGF-β type 1 and 2 receptors. A single case report of a patient who improved level of activity during Losartan therapy suggests that downregulation of TGF-B1 receptor may be an approach to decrease the effects of high levels of TGF-B. Surgical decompression of the narrowed foramina may improve the cranial nerve palsies. In sclerosteosis there is no effective medical therapy. Bony syndactyly is difficult to repair. Neurologic symptoms from cranial nerve root compression and the subsequent complications are managed surgically.
Melorheostosis
Melorheostosis is Greek for “flowing hyperostosis,” which is taken from the radiographic appearance of the involved bone that resembles melted wax that has dripped down a candle. The rare disease is an acquired sclerosing bone disorder of unknown etiology.
Clinical
The major manifestation is progressive linear hyperostosis in one or more bones of one limb, usually a lower extremity. If bilateral, the disease is usually asymmetric. Symptoms appear in childhood as pain and/or stiffness in the area of the sclerotic bone with the lesions advancing rapidly. Associated soft tissue masses may be present and include scleroderma-like features. Other changes that may overlie the bone lesions include hypertrichosis, ectopic cartilage, bony tissue, fibromas, and hemangiomas. These superficial lesions may become evident before the underlying skeletal changes. In adults, pain and stiffness may persist, and there may be contractures of nearby joints, but the skeletal lesions do not progress.
Histology
The skeletal changes of melorheostosis are endosteal thickening during childhood and periosteal new bone formation during the adult years. There is sclerosis with thick and irregular lamellae. Examination of the skin overlying the skeletal lesions may contain a scleroderma-like lesion, but the composition of collagen is normal and different from collagen in scleroderma lesions.
Radiography
The changes characteristic of melorheostosis involve a single bone or an occasional adjacent bone with irregular, dense hyperostosis involving both periosteal and endosteal surfaces ( Fig. 12.4 ). Bones of the lower extremities are most commonly involved, but the lesions have been found in other bones as well. Ectopic bone may be present in the soft tissue in proximity to the skeletal lesions and near joints. Nuclear medicine bone scans show accumulation of tracer in the lesions indicating high blood flow and can be useful in differentiating melorheostosis from other osteopoikilosis and osteopathic striata.
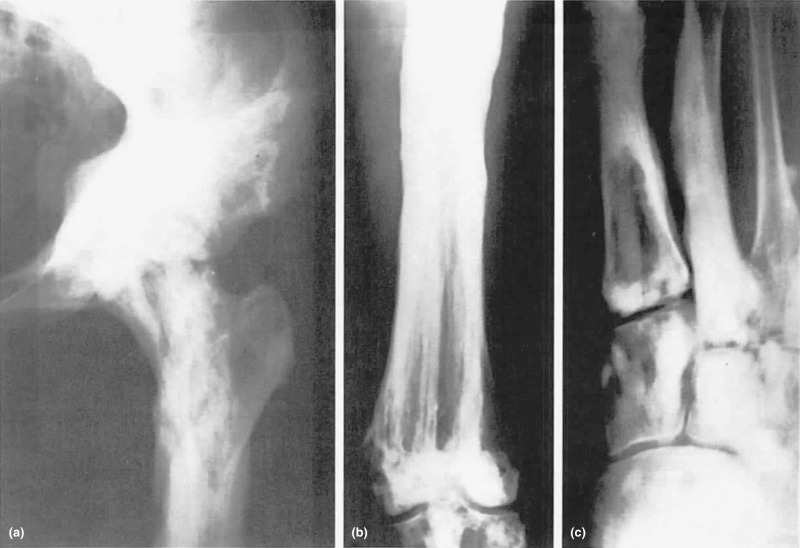
Laboratory
Laboratory tests including serum calcium, phosphorus, and alkaline phosphatase are normal.
Genetic Pathophysiology
No specific etiology is known. A small number of patients with a mutation in the LEMD3 gene that causes poikilosis have some phenotypic changes suggestive of melorheostosis, but the majority of melorheostosis patients are sporadic and have no LEMD3 abnormality. A hypothesis that has not been tested is that the disease arises from a somatic mutation confined to the affected bone.
Genetic Diagnostic Testing
There is no available genetic testing available. Radiotracer scanning of the skeleton will demonstrate increased uptake of a single bone consistent with that involved on standard radiography.
Treatment
No specific treatment is available. Surgical repair of contractures is often unsuccessful due to recurrence of the deformities.
High Bone Mass Phenotype
Selective activating mutations of the LRP5 gene, which encodes for low-density lipoprotein receptor-related protein 5, is inherited as an autosomal dominant trait with the major finding of a systemic increase in skeletal mass composed of normal bone. Serum sclerostin is elevated in men and women with high bone mass with or without an hereditary LRP5 activating mutation. The high bone mass is associated with an increased Wnt signaling and stimulation of osteoblasts. Therefore, these observations indicate there is resistance to sclerostin’s inhibitory actions on bone formation. Other features of the phenotype may be observed including torus palatinus, cranial nerve palsies, and oropharyngeal exostoses. Detection of the lesions in other family members is done by finding high DEXA scanning and dense radiographic imaging of young adults at risk.
Juvenile Paget’s Disease
The disorder, also known as hereditary hyperphosphatasia or osteoectasia with hyperphosphatasia, is characterized clinically by infants and children with progressive bone deformities, fractures, short stature, and elevated bone alkaline phosphatase. Families have been characterized as having a homozygous deletion of the TNFRSF11B gene, which encodes for the circulating receptor activator of nuclear factor-kB (RANKL)-binding protein osteoprotegrin. The familial pattern of disease is consistent with an autosomal dominant pattern of inheritance with variable penetrance. Radiographic findings are the result of uncontrolled, high rates of osteoclastic resorption ( Fig. 12.5 ).
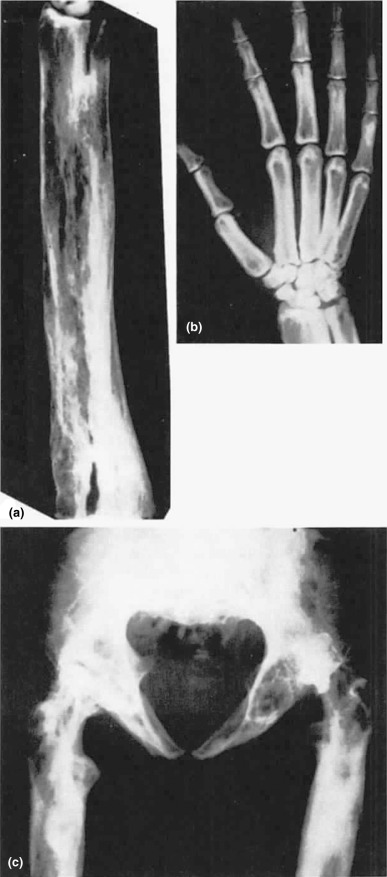
A rare Paget’s disease-like disorder called familial expansile osteolysis is an autosomal dominant condition marked by accelerated bone remodeling has been mapped to chromosome 18q21-22. Mutations have been located to exon 1 of the TNFRSF11A gene, which encodes for RANK, a pivotal molecule in the activation of osteoclastic bone resorption. In other families, susceptibility loci have been mapped to a number of chromosomes resulting in mutations of TNFSF1 (RANKL), TNFRS11A (RANK), and TNFRSF11B (OPG), which explain the phenotypic similarities with the Paget’s disease phenotype.
Treatment of these related disorders has been antiresorptive agents, including intravenous bisphosphonates and denosumab.
Osteopathia Striata
Osteopathia striata with cranial sclerosis (OSCS) is an X-linked dominant condition marked by linear striations, mainly affecting the metaphyseal region of the long bones including those of the hands, feet, and pelvis. Recently, the disease-causing gene was identified as the WTX gene, an inhibitor of WNT signaling. Skeletal changes are usually benign but in some patients may be accompanied by cranial sclerosis due to a mutation of the WTX gene. The WTX protein functions as a tumor suppressor and inactivating mutations have been associated with some cancers. However, no increase in cancer among patients with osteopathia striata has been reported.
Females demonstrate sclerotic striations on the long bones, cranial sclerosis, and craniofacial dysmorphism. Males have significant skeletal sclerosis, do not have striations but do display a more severe phenotype commonly associated with gross structural malformations, patterning defects, and significant pre- and postnatal lethality. A more serious variant with high lethality is notable for the presence of focal dermal hypoplasia with bony limb defects in males with WTX mutation and transmission as an X-linked recessive trait. Diagnosis is made from the radiographic appearance of the lesions. Genetic testing of a WTX mutation would be supportive of the diagnosis, but the absence of a WTX mutation does not exclude the disease. There are no specific laboratory tests that are characteristic of the disorder. No specific therapy is known.
Osteopoikilosis
The disease of “spotted bones” is a benign autosomal dominant disorder free of symptoms and discovered by its radiologic changes. Osteopoikilosis can occur either as an isolated anomaly or in association with other abnormalities of skin and bone.
Radiographic Findings
The lesions are characterized by a symmetric but unequal distribution of multiple hyperostotic areas in different parts of the skeleton. Small, numerous, round or oval foci of bony sclerosis are seen in the epiphyses and adjacent metaphyses. The lesions are mainly in long bones and not in the skull, vertebrae, or ribs.
Histology
The lesions represent foci of mature remodeled bone with lamellar structure, either connected to adjacent trabeculae of spongy bone or attached to the subchondral cortex.
Genetic Findings
A genome-wide linkage analysis followed by the identification of a microdeletion in an unrelated individual with these diseases allowed a map of the gene that is mutated in osteopoikilosis. In one series, all the affected individuals investigated were heterozygous with respect to a loss-of-function mutation in LEMD3 (also called MAN1 ), which encodes an inner nuclear membrane protein. A novel C to T substitution at position 2032 bp (cDNA) in exon 8 of LEMD3 , resulting in a premature stop codon at amino acid position 678, has been described. The mutation cosegregates with the osteopoikilosis phenotype in two other reported families. Often confused with metastatic lesions, the osteopoikilosis lesions are stable over time and do not take up radiotracer of nuclear bone scanning. No treatment is indicated.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


