Abstract
Growth hormone (GH) insensitivity can result from molecular defects at multiple levels, including the genes for the GH receptor ( GHR ), signal transducer and activator of transcription ( STAT )5B, insulin-like growth factor-I ( IGF1 ), the acid-labile subunit carrier protein for IGF ( IGFALS ), or the IGF-I receptor ( IGF1R ). This chapter summarizes molecular defects at each of these levels. A database and website for all published cases is available at www.growthgenetics.com .
Keywords
short stature, Turner syndrome, intrauterine growth failure, GH receptor ( GHR ), IGF-I, STAT5b, IGF 1 receptor
Introduction
Background, Incidence, Prevalence
For starters, short stature is not a disease; nor, for that matter, is growth failure. However one chooses to define short stature, it is, ultimately, a statistical definition, based (probably fallaciously) on the assumption that stature follows a perfect Gaussian distribution. Accordingly, 3% of children (or adults) fall below the third percentile of stature, and approximately 1.2% fall below −2.25 standard deviations, the FDA-approved definition of “idiopathic short stature.”
It is stated above that stature is not likely to follow a perfect bell-shaped curve because it is clear that there are many more pathological conditions that result in growth failure than in overgrowth. Many chronic diseases of childhood (e.g., chronic renal failure, inflammatory bowel disease, cystic fibrosis, rheumatoid arthritis, immunodeficiency states, chronic infection, etc.) are characterized by growth failure. The alert clinician recognizes that short stature may be an important symptom of underlying diseases outside of the endocrine system and, when appropriate, short patients should be evaluated for such conditions.
Additionally, many chromosomal disorders are characterized by short stature, as commonly observed in Turner syndrome (45,X or various abnormalities of the short arm of the X chromosome) and trisomies 21, 13, and 15. To such conditions, one may add a wide range of chromosomal deletions, inversions, or translocations. These pathologic states are characterized, generally, by characteristic dysmorphic features, which will help steer the clinician in the direction of a chromosomal anomaly, but short stature may, at times, be the presenting sign. A good general rule, for example, is that any female with unexplained short stature warrants a karyotype to rule out Turner syndrome, even in the absence of some of the typical signs or symptoms. Similarly, when short stature is associated with dysmorphic features and/or intrauterine growth failure, the possibility of a chromosomal defect should be excluded.
Any evidence of disproportionate short stature raises the question of an inborn skeletal dysplasia. In the 2010 revision of the International Nosology and Classification of Genetic Skeletal Disorders, 456 conditions, divided among 40 groups, were defined by molecular, biochemical and/or radiographic criteria ( http://isds.ch/uploads/pdf_files/Nosology2010.pdf ). Some of these conditions result in drastic growth failure, and frequently, early mortality. Others can be relatively subtle and may not be evident without careful ascertainment of skeletal proportions, as well as radiologic evaluation of the long bones, spine, extremities, and skull. While specific mutations have now been identified for many of these conditions, and diagnostic tests are increasingly available, a discussion of skeletal dysplasias is beyond the scope of this chapter. An International Skeletal Dysplasia Registry has been created, and the reader is directed to an excellent review by Rimoin et al. and to the website www.csmc.edu/skeletaldysplasia .
Disturbances in each of two hormonal systems may result in profound growth failure. Chronic hypothyroidism, either primary or secondary, may lead to severe stunting of growth, and all children with significant growth failure should have their thyroid status evaluated. Growth hormone (GH) deficiency, on either a hypothalamic or pituitary basis, can also result in dramatic postnatal growth failure, and is the subject of Chapter 7 (Congenital Defects of Thyroid Hormone Synthesis).
This chapter focuses on genetic defects of the GH-insulin-like growth factor (IGF) axis, distal to the production of bioactive GH. As such, it encompasses defects of the GH receptor (GHR), the GH signaling cascade, the genes for IGF-I and IGF-II, the IGF binding proteins, the IGF-I receptor (IGF1R), and the structurally-related insulin receptor (IR), and signaling defects of either (or both) the IGF1R and the IR. The combination of targeted animal gene knockout studies and human mutational analysis has unequivocally demonstrated the critical and major roles that the IGF system plays in mammalian growth. In murine models, approximately two-thirds of total prenatal and postnatal growth is IGF dependent, and the preponderance of the growth-promoting actions of GH are attributable to its stimulation of IGF-I gene transcription. In light of the central role of IGF-I in mammalian postnatal growth, and by analogy with other pituitary-based endocrine systems, it is logical to divide IGF deficiency (IGFD) into secondary and primary forms: “secondary IGFD” encompasses IGF deficiency resulting from a failure of production or secretion of bioactive GH on either a hypothalamic or pituitary basis (see Chapter 7 ); “primary IGFD” encompasses IGF deficiency, existing in the presence of normal or elevated GH production or serum concentrations (see Table 26.1 ).
|
The prevalence of primary IGFD is difficult to assess; as is often the case, ascertainment bias, a significant factor in dealing with the phenotype of short stature, enters into the picture. To date, the total number of reported cases of the conditions listed in Table 26.1 is approximately 300−400, with the majority reflecting genetic abnormalities of GHR . It is apparent, however, that such cases are likely to represent the tip of the iceberg. It has been estimated, for example, that 25−40% of children with short stature (defined as heights less than −2.25 SD for age) have serum concentrations of IGF-I below −2 SD. The molecular basis for these observations is unclear at this time, although the subject of active investigation.
Homozygous disruption of IGF1R in mice resulted in severe intrauterine growth failure and early demise. To date, the majority of reported patients with IGF1R mutations have defects limited to one allele (heterozygous). Several patients have been described, however, with compound heterozygous or homozygous IGF1R defects. While such patients should not be described as IGF deficient, their growth failure further confirms the critical role of the IGF axis in prenatal and postnatal growth failure.
The IGFD Research Center, established in 2006 and located in Portland, Oregon, has served as a worldwide referral center for patients with unexplained growth failure and potential molecular defects resulting in primary IGFD or IGF resistance. The Center has received approximately 75−100 cases annually, from over 25 different countries, and has identified novel molecular defects at all levels of the GH-IGF axis described in the subsequent sections. The IGFD Research Center is now part of a coordinated international effort to construct, develop, and curate an international database and website for molecular defects of the GH-IGF axis.
Clinical Presentation
Short stature is the defining characteristic, naturally, but defining short stature can be challenging. When growth failure commences in utero , intrauterine growth retardation (IUGR) occurs, resulting in newborns who are small for gestational age. Intrauterine growth failure must be differentiated from postnatal growth failure, as the two conditions may or may not accompany one another.
As in the case of GH deficiency, GH insensitivity, either at the level of the GHR or the postreceptor GH signaling cascade, is characterized, typically, by relatively normal intrauterine growth, but severe postnatal growth failure, commencing within the first few months of life. Mutations or deletions of the IGF1 gene, or mutations affecting IGF1R , on the other hand, combine intrauterine growth retardation with poor postnatal growth. Thus, while GH does not appear to be essential for intrauterine growth, IGF-I itself is critically involved in both prenatal and postnatal growth. Presumably, IGF-I production in utero is largely GH independent, but switches to profound GH dependence sometime near birth. Defects of IGF1 are also characterized by microcephaly, developmental delay and, in some cases, sensorineural deafness.
In addition to postnatal growth failure, patients with GHR defects have infantile facies, cephalofacial disproportion, truncal adiposity, bluish sclerae and delayed dentition, and skeletal maturation, much as is commonly observed in GH deficient children. A tendency to fasting hypoglycemia is observed in patients with either GHD or GHI, presumably reflecting the loss of counterregulatory actions of GH in glucose metabolism.
Table 26.2 displays characteristic biochemical findings of patients with defects of the GH-IGF axis. The combination of clinical phenotype and biochemical findings generally can helps direct the clinician toward the most appropriate molecular diagnoses to consider. While many of the features of these conditions overlap, each molecular defect carries characteristic biochemical and clinical features, resulting in a continuum of findings.

Genetic pathophysiology
Known Mutations and Specific Mutations
Mutations/Deletions of GHR
Abnormalities of the GHR constitute both the earliest and the most prevalent identified molecular defects resulting in primary IGFD. Although the original genetic abnormalities reported were large deletions of GHR , involving exons 3, 5, and 6 (we now know that exon 3 may be spliced out in approximately one-third of the normal population), the majority of gene abnormalities have turned out to be point mutations. The reported deletions of exons 5 and 6 lead to a frameshift, with a consequent premature translational stop signal in exon 7 and a resultant aberrant protein lacking both the transmembrane and intracellular domains.
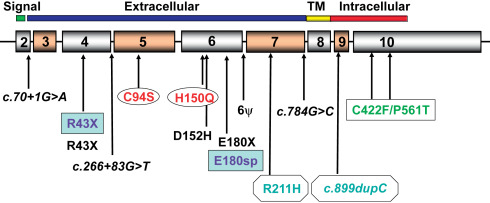
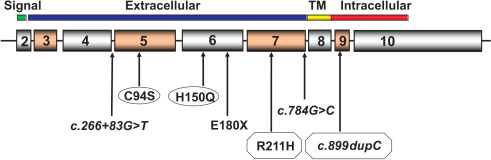
At least 70 mutations of GHR have been identified to date, including missense, nonsense, frameshift, and splice defects. These mutations are summarized in a recent review. Figs. 26.1 and 26.2 show homozygous and compound heterozygous mutations of GHR identified in the IGFD Research Center. Approximately 90% of these involve the extracellular, GH-binding domain of the GHR and almost all are characterized by reduced serum concentrations of GH binding protein (GHBP). Of interest is an intronic base change that results in activation of a pseudoexon sequence of 108 nucleotides and the insertion of 36 new amino acids within the extracellular domain of the GHR. Phenotypes associated with this pseudoexon activation are highly variable and include patients whose stature and serum IGF-I concentrations fall within the lower portion of the normal range.
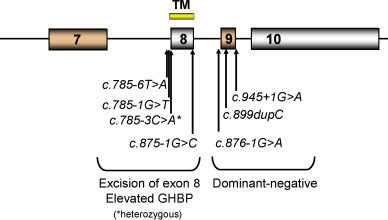
Perhaps of greater interest are the rarer cases of abnormalities involving the transmembrane or intracellular domains of the GHR, as these patients have normal or even elevated serum concentrations of GHBP and may not, consequently, be initially identified as having GHR abnormalities ( Fig. 26.3 ). A heterozygous C→A transversion at position c.785-3 at the acceptor site of intron 7 results in the excision of exon 8 of the GHR , impaired anchoring of the receptor to the cell membrane, and markedly elevated serum GHBP. Three splice-site mutations in exon 9 have been reported to behave in a dominant negative manner. This has been attributed to the production of truncated GHR molecules, lacking most of the intracellular domain, but retaining extracellular and transmembrane domains, allowing them to dimerize with normal GHR molecules and, thereby, perturb GH signaling. Three deletions in exon 10 have been identified: 309delC results in GHR 1-330; a 22bp deletion results in GHR 1-449; and a 1776delG leads to a GHR molecule with the first 560 amino acids, followed by a nonsense sequence from 560 to 581. A number of polymorphisms and potential heterozygous missense mutations of the intracellular domain have been identified, but their functional significance remains uncertain at this time.