cancers.6 Because it is not feasible to accurately predict whether a particular patient’s tumor will respond to a given drug, administering anticancer drugs in combination ensures a greater chance of achieving a response (i.e., exposing the tumor to at least one active agent). In addition to providing a broader range of coverage against naturally resistant tumor cells, combination chemotherapy may also prevent or delay the development of acquired resistance in initially responsive tumors and provide additive or synergistic cytotoxic effects if agents with different mechanisms of action are selected.
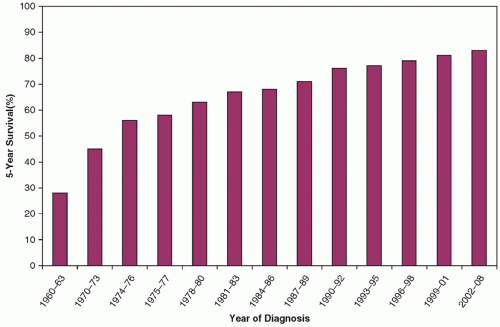 Figure 10.1 Five-year survival rate for all childhood cancers diagnosed between 1960 and 2004.(From Silverberg E, Boring CC, Squires TS. Cancer statistics, 1990. CA Cancer J Clin 1990;40(1):9-26; Jemal A, Murray T, Ward E, et al. Cancer statistics, 2005. CA Cancer J Clin 2005;55:10-30; Jemal A, Siegel R, Ward E, et al. Cancer Statistics, 2009. 2009;59(4):225-249.) |
This approach, called primary or neoadjuvant chemotherapy, may also improve local control of the primary tumor by shrinking the primary and making it more amenable to surgical resection, in addition to providing earlier therapy for micrometastases.18
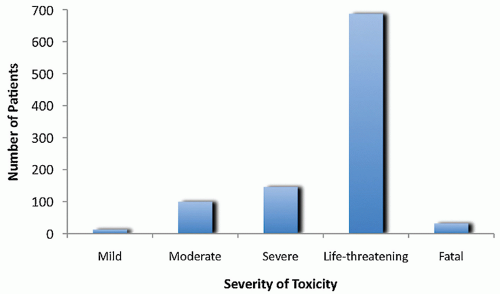 Figure 10.2 The worst degree of any toxicity experienced by patients (n = 1,062) treated on one of the eight treatment arms of the Intergroup Rhabdomyosarcoma Study III. Seventy-eight percent of patients had at least one severe or life-threatening toxicity, and there were thirty-two toxicity-related deaths. (Adapted from Table 6 in Crist W, Gehan EA, Raghab AH, et al. The third Intergroup Rhabdomyosarcoma Study. J Clin Oncol 1995;13:610.) |
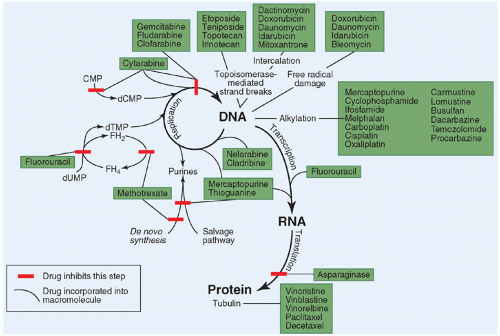 Figure 10.3 Site of action of the commonly used cytotoxic anticancer drugs. |
interrelated pathways can result in loss of control of DNA replication and cell division and suppression of the apoptotic response to receptor-linked or DNA-damage-induced signals. In addition to their role in tumorigenesis, mutations in cell cycle regulatory genes and genes involved in apoptosis may modulate the sensitivity of cancer cells to conventional anticancer drugs.40,42 The cellular damage produced by most anticancer drugs appears to induce apoptosis in chemosensitive cancer cells. Overexpression of oncogenes that promote apoptosis, such as MYCC and MYCN, can enhance the chemosensitivity of tumor cells, whereas overexpression of bcl-2, which blocks the apoptotic pathway, can attenuate drug-induced apoptosis and convey pleiotropic resistance to anticancer drugs.
TABLE 10.1 Pharmacokinetic Terms | ||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||
As the technology to measure the concentration of these drugs and their metabolites in biologic fluids has improved, a greater emphasis has been placed on studying anticancer drug pharmacokinetics in children with cancer.
toxicities must be tolerated to administer effective doses.95 Acute toxicities common to many of the anticancer drugs include myelosuppression, nausea and vomiting, alopecia, orointestinal mucositis, liver function abnormalities, allergic or cutaneous reactions, and local ulceration from subcutaneous drug extravasation. These acute toxicities occur over hours to weeks after a dose and are usually reversible. Many drugs also have unique toxicities affecting specific organs or tissues, such as cardiotoxicity associated with the anthracyclines, hemorrhagic cystitis associated with cyclophosphamide and ifosfamide, peripheral neuropathy from vincristine, cisplatin, and paclitaxel, nephrotoxicity from cisplatin and ifosfamide, ototoxicity from cisplatin, and coagulopathy from L-asparaginase. Many of these latter toxicities are cumulative (i.e., occur after multiple doses), and in some cases they are not completely reversible (e.g., anthracycline cardiotoxicity).
TABLE 10.2 Physiologic Differences in Children that May Influence Drug Disposition | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
doses to avoid overlapping toxicities. For example, nonmyelosuppressive agents such as vincristine, prednisone, L-asparaginase, and high-dose methotrexate with leucovorin rescue can often be administered with traditional myelosuppressive drugs without compromising the dose of myelosuppressive agents. Some regimens administer nonmyelosuppressive agents during the period of marrow suppression from myelotoxic drugs to ensure continuous exposure of the tumor to cytotoxic therapy.107
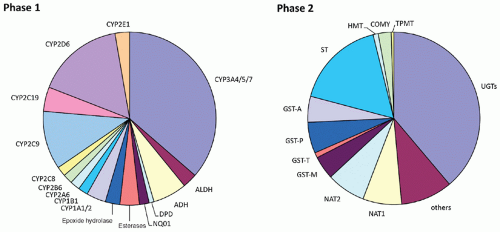 Figure 10.4 Phase I and II enzymes involved in drug metabolism. Almost all of the major human enzymes responsible for modification of functional groups or conjugation with endogenous substituents exhibit common polymorphisms at the genomic level. The percentage of phase I and phase II metabolism of drugs that each enzyme contributes is estimated by the relative size of each section of the corresponding chart. ADH, alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; CYP, cytochrome P450; DPD, dihydropyrimidine dehydrogenase; NQO1, NADPH:quinone oxidoreductase or DT diaphorase; COMT, catechol-O-methyltransferase; GST, glutathione S-transferase; HMT, histamine methyltransferase; NAT, N-acetyltransferase; STs, sulfotransferases; TPMT, thiopurine methyltransferase; UGTs, uridine 59-triphosphate glucuronosyltransferases. (Adapted with permission from Evans WE, Relling MV. Pharmacogenomics: translating functional genomics into rational therapeutics. Science 1999;286(5439):487-491. Copyright 1999 AAAS) |
as the gene amplification identified in methotrexate-resistant cells that results in overproduction of dihydrofolate reductase (DHFR), the target enzyme of methotrexate.115 Mechanisms of resistance to molecularly targeted receptor and nonreceptor TKIs include point mutations in the kinase domain that lower drug affinity or activation of downstream effectors or alternative signaling pathways through epigenetic alterations.116,117
the childhood cancers. The chemical structures of these agents and several nonclassical alkylators are shown in Figures 10.6, 10.7, 10.8, and 10.9.
TABLE 10.3 Pharmacologic Properties of the Commonly Used Anticancer Drugs | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 10.4 Pharmacokinetic Parameters of the Commonly Used Anticancer Drugs | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
most widely used in pediatric oncology (Fig. 10.6). Even with this long history, new nitrogen mustards such as bendamustine continue to be developed in treatment regimens for cancer.
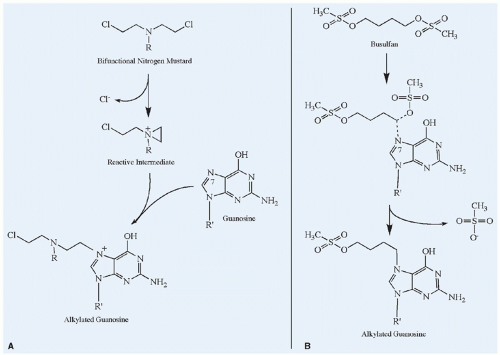 Figure 10.5 Mechanisms of alkylation of the nucleophilic N7 position of guanosine. A: The bifunctional nitrogen mustard illustrates the SN1 type of alkylation reaction in which a reactive intermediate forms spontaneously and then rapidly reacts with the nucleophilic group. The rate-limiting step for SN1 alkylation is the formation of the reactive intermediate, and thus the reaction exhibits first-order kinetics (i.e., independent of the target nucleophile concentration). If the second chloroethyl group also reacts with another nucleotide base, a cross-link is formed. B: Busulfan exemplifies an SN2 reaction, characterized by a bimolecular nucleophilic displacement. In this case, the methylsulfonate group on either end of busulfan is displaced by the nucleophilic group on guanosine. The rate of SN2 alkylation reactions depends on the concentration of the alkylating agent and the target nucleophile, and it therefore follows second-order kinetics. |
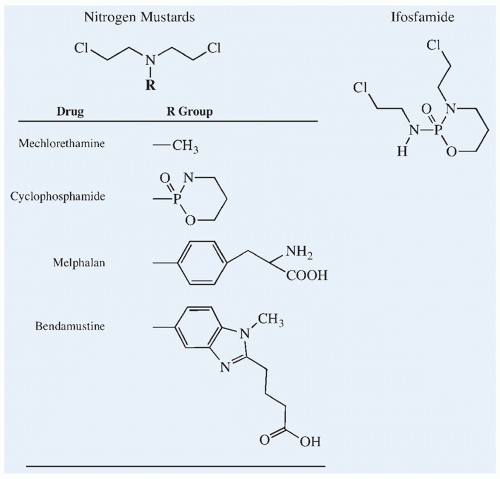 Figure 10.6 Chemical structures of the nitrogen mustard alkylating agents and the cyclophosphamide isomer, ifosfamide. |
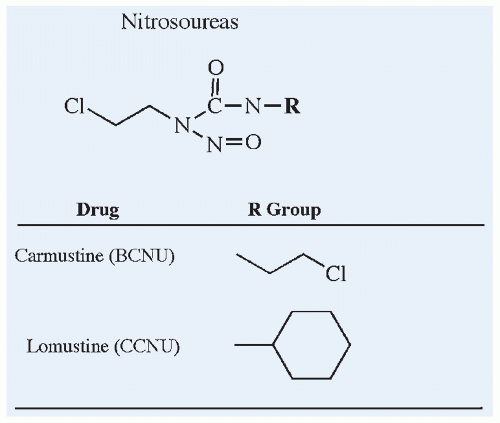 Figure 10.7 Chemical structures of the nitrosoureas, carmustine and lomustine. |
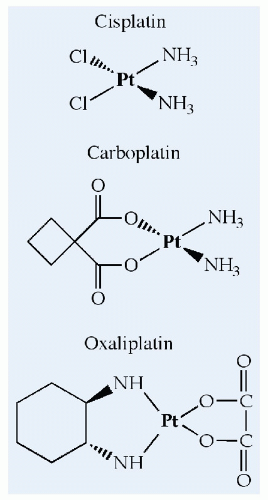 Figure 10.8 The chemical structures of cisplatin, carboplatin, and oxaliplatin, which platinate DNA in a manner analogous to alkylation by the nitrogen mustards. Reactive intermediates are formed after spontaneous elimination of chloride (cisplatin), dicarboxylate cyclobutane (carboplatin), or oxalate (oxaliplatin). |
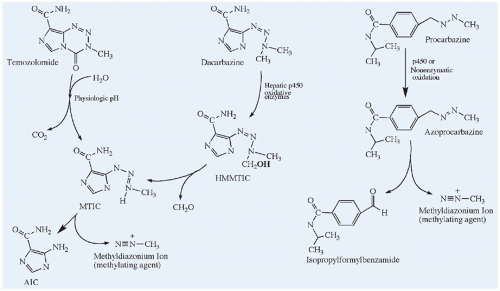 Figure 10.9 Chemical structures and activation pathways of the methylating agents, dacarbazine, temozolomide, and procarbazine, which are prodrugs. Dacarbazine requires enzymatically catalyzed activation, and temozolomide undergoes spontaneous chemical conversion in solution at physiological pH to the active metabolite, MTIC. MTIC, methyltriazenyl-imidazole carboxamide; HMMTIC, hydroxymethyl-MTIC; AIC, amino-imidazole carboxamide. The metabolic pathway for procarbazine is highly complex and incompletely shown. In addition to the methyldiazonium ion, free radicals can also be generated from azoprocarbazine. |
was not studied in the United States until the 21st century, and was approved in 2008 by the U.S. FDA for the treatment of indolent B-cell lymphoma.156,157
 Figure 10.10 Metabolic pathways for the oxazaphosphorines, cyclophosphamide and ifosfamide. Both compounds must undergo hydroxylation at the 4 position before expressing alkylating activity; this reaction is catalyzed by hepatic microsomal enzymes. The 4-hydroxy metabolites are in spontaneous equilibrium with the open-ring aldehydes (aldophosphamide or aldoifosfamide), which can release Acrolein and form the active alkylating mustards (phosphoramide mustard or isophosphoramide mustard). Further oxidation at the 4 position of the primary metabolites leads to the formation of inactive metabolites (ketocyclophosphamide and carboxyphosphamide or ketoifosfamide and carboxyifosfamide), which are excreted in the urine. The open-ring aldehyde metabolites can be chemically reduced to an alcohol (alcophosphamide or alcoifosfamide). Inactivation by dechlorethylation leads to formation of the potentially toxic by-product chloracetaldehyde. This is a minor pathway for cyclophosphamide but more active with ifosfamide. |
which are formed by spontaneous elimination of acrolein from the open-ring aldehydes. Quantitatively, the rate of activation of cyclophosphamide is greater than that of ifosfamide, and this difference in the rate of activation accounts for the difference in clinical pharmacokinetics and MTD of the two isomers.167,168,169
in water retention.213,214 Ifosfamide produces proximal tubular damage resembling Fanconi syndrome, with glucosuria, amino aciduria, and phosphaturia. Animal studies suggest that it is the ifosfamide metabolite chloracetaldehyde, acting on mitochondrial NADH:ubiquinone oxidoreductase in the renal tubule, which is the primary mediator of nephrotoxicity.215 Rickets has been observed in younger children.216,217,218,219,220 Decreased glomerular filtration rate (GFR) and distal tubular damage manifested by concentrating defects and renal tubular acidosis also has been reported.221,222 Comprehensive follow-up evaluation of glomerular and tubular function in children previously treated with ifosfamide revealed dysfunction in 78%, including 28% with moderate or severe nephrotoxicity.223 Cumulative doses of 45 to 80 g/m2 or greater appear to be the primary risk factor,223,224,225 with young children appearing to be at higher risk for proximal renal tubular damage.226,227
leukemia are late effects associated with the chronic administration of melphalan. At high doses with autologous bone marrow or stem cell reinfusion, gastrointestinal toxicity (e.g., mucositis, esophagitis, diarrhea) becomes dose limiting.141,251,268
many bone marrow transplant preparative regimens, usually in combination with cyclophosphamide.140
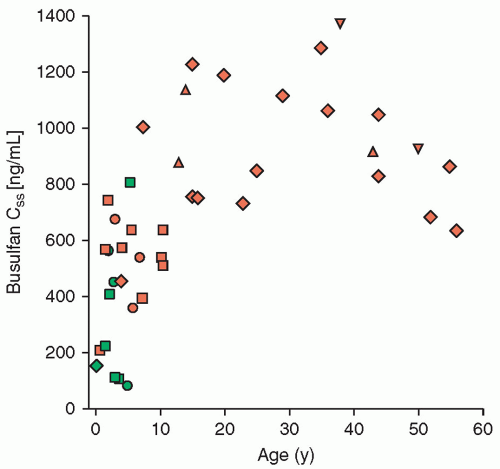 Figure 10.11 Plasma busulfan steady-state concentrations (Css) as a function of age. Css is derived by dividing the AUC by the dosing interval (6 hours). Patients were treated with 16 to 30 mg/kg of busulfan in combination with cyclophosphamide prior to bone marrow transplant. Triangles represent patients who rejected their graft or had a mixed chimera. Patients who experienced grade 0 treatment-related toxicity are designated in green, grade 1 toxicity in dark blue, grade 2 toxicity in orange, grade 3 toxicity in red, and grade 4 toxicity in black. Young children had substantially lower Css, less toxicity, and were at greater risk for graft rejection. (From data presented in Tables 1, 2, and 3 in Slattery JT, Sanders JE, Buckner CD, et al. Graft rejection and toxicity following bone marrow transplantation in relation to busulfan pharmacokinetics. Bone Marrow Transpl 1995;16:31-42.) |
5 days. The divided dose and continuous-infusion schedules may lessen the gastrointestinal and renal toxicities.344,348 Cisplatin has been administered regionally in a number of trials, including intraperitoneally for ovarian cancer, intravesicularly for bladder cancer, intrapleurally for the malignant pleural effusions, and intra-arterially for brain tumors and for sarcomas of the extremity, including osteosarcoma.349,350
must be exercised when using these formulas, however, as the results are expressed either as an absolute dose (mg) or as a dose normalized to body surface area (mg/m2).389 When administered as a single dose in combination with ifosfamide and etoposide, a targeted carboplatin AUC of up to 10 mg • min/mL was tolerable.388 In ovarian and testicular cancers in adults, a carboplatin AUC of 5 to 7 mg • min/mL was associated with a higher response rate and a lower risk of disease recurrence.384 Additional study of the pharmacokinetics of carboplatin in infants is needed, as an analysis of carboplatin exposures achieved in patients less than 1 year of age suggests that clearance of this drug may be different in very young patients.390
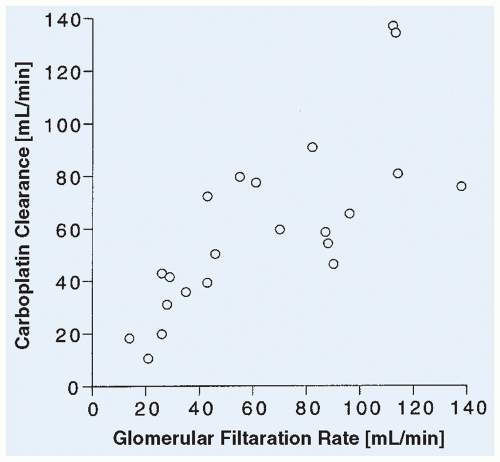 Figure 10.12 Relation between carboplatin clearance and GFR as measured by 51Cr-EDTA clearance in 22 children. (Adapted from Newell DR, Pearson ADJ, Balmanno K, et al. Carboplatin pharmacokinetics in children: the development of a pediatric dosing formula. J Clin Oncol 1993;11:2314.) |
TABLE 10.5 Adaptive Dosing Formulas for Targeting Carboplatin Dose to Achieve a Desired Nadir Platelet Count or AUC, with the Target AUC Ranging from 7 to 10 mg • min/mL | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||
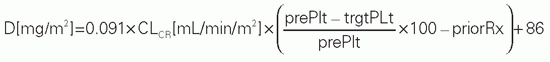
doses of 300 to 600 mg/m2.229,393 Lhermitte sign (an electric shock sensation when the neck is flexed) is common at high cumulative doses of cisplatin.408 Symptoms may progress after discontinuation of cisplatin and persist for months to years. Seizures and encephalopathy have also been reported in children receiving intensive cisplatin therapy.409 The irreversible hearing loss is in the high-frequency range and appears to be related to a cumulative dose of cisplatin of greater than 400 mg/m2.410,411,412 Children under 5 years of age also appear more likely to develop cisplatin-related hearing loss compared with older children.413 Genetic differences may explain some of the variability in platinum-associated neurotoxicity in adults, though the significance of germline variants in genes such as TPMT and catechol-O-methyltransferase (COMT) is a subject of debate in the literature.414,415,416 Amifostine decreases the incidence and severity of platinum-related neurotoxicity and ototoxicity in adults.417 In a study of children with average-risk medulloblastoma, amifostine appeared to have provided some otoprotection,418 but protective effects have yet to be observed in other pediatric studies.419,420,421 More recently, sodium thiosulfate has been studied as an otoprotectant.422 Although it appears to lessen the ototoxicity of cisplatin, the lack of selectivity appears to confer a tumor-protective effect that may curtail further clinical development. Additional toxic effects associated with cisplatin include prominent nausea and vomiting, mild myelosuppression, Raynaud phenomenon, and hypersensitivity reactions.344
schedule included a total clearance of 110 mL/min/m2, a volume of distribution at steady state of 23 L/m2, and a terminal half-life after infusion of 3 hours.
that results from administering these agents by continuous infusion or by chronic daily dosing increases the chance of exposing a higher proportion of the tumor cell population to the drugs during active DNA replication.
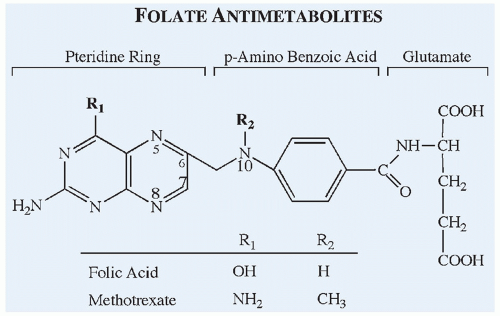 Figure 10.13 Chemical structures of the antifolate methotrexate compared with the structure of folic acid. |
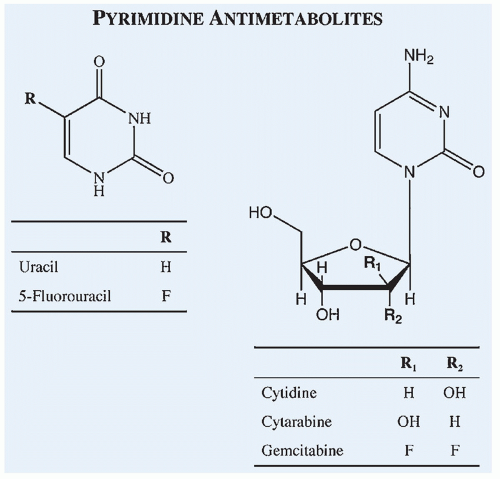 Figure 10.14 Chemical structures of commonly used pyrimidine antimetabolites compared with the structures of corresponding endogenous compounds of which they are analogs. |
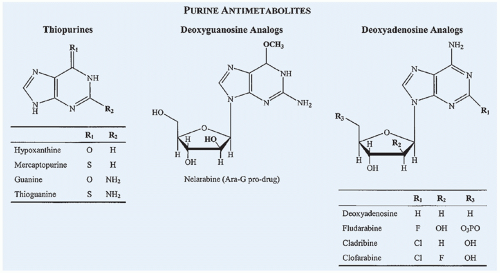 Figure 10.15 Chemical structures of commonly used purine antimetabolites compared with the structures of corresponding endogenous compounds of which they are analogs. |
single-carbon group and is oxidized to dihydrofolate in the conversion of deoxyuridylate (dUMP) to thymidylate (dTMP) by thymidylate synthase. In the presence of methotrexate, intracellular tetrahydrofolate pools are depleted, leading to depletion of purines and thymidylate and inhibition of DNA synthesis. Accumulation of partially oxidized dihydrofolic acid, resulting from the inhibition of DHFR, appears to contribute to the inhibition of de novo purine synthesis.471,472 A critical determinant of methotrexate cytotoxicity is the rate of thymidylate synthesis, because the synthesis of thymidylate from uridylate is the only reaction that oxidizes the tetrahydrofolate cofactor to the inactive dihydrofolate form. Another determinant is achieving an intracellular methotrexate concentration that is in excess of DHFR-binding sites, because intracellular levels of this target enzyme are 20-fold to 30-fold higher than that required to maintain tetrahydrofolate pools.470,473,474
significant toxicity. These toxicities can be prevented by administration of leucovorin. With the use of therapeutic drug monitoring and continuation of leucovorin rescue until plasma methotrexate concentration has fallen below 0.05 to 0.1 µM, the toxicity of high-dose methotrexate can be avoided in most patients.469 Despite these measures, however, nephrotoxicity still occurs in almost 2% of patients receiving HDMTX infusions.516
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


