TABLE 29.1 Principal Histologic Types of Soft Tissue Sarcoma in the World Health Organization (WHO) Classification of Bone Tumors with International Classification of Diseases for Oncology (ICD-O) Morphology Rubrics Listed in Alphabetic Order | ||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||
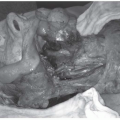
 FIGURE 29-1. Hemangiopericytoma of the left nasal fossa in a 40-year-old. This patient underwent resection with positive margins for well circumscribed lesion in 1994 (A and B). Postoperative radiotherapy to a dose of 60 Gy achieved durable local control. After 8 years, he relapsed with two bone lesions one in the left chest wall and the second in the right pelvis without other lesions shown on nuclide bone scan (C). Both lesions were successfully treated with combined radiotherapy and surgery but diffuse metastases resulted several years later. The original primary site also shows nucleotide uptake but cross-sectional anatomic imaging and clinical examination failed to reveal recurrent tumor. |
TABLE 29.2 Cytogenetic Abnormalities in Soft Tissue Sarcoma | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
parameters (differentiation score, mitoses, necrosis), but its greatest limitation lies in the assignment of a differentiation score.13 Roughly defined as the extent to which a lesion resembles normal tissue, differentiation score has little applicability for tumors which ostensibly have no normal tissue counterpart.
 FIGURE 29-2. A: Coronal projection of computed tomography (CT) of an 18-year-old patient with synovial sarcoma arising from the left temporomandibular joint region. There is a large heterogeneously enhancing soft tissue mass causing distortion of the facial contour. Remodeling of bone is apparent as well as spiculated periosteal reaction and calcification. The diagnosis was established by the characteristic molecular gene fusion and translocation seen in synovial sarcoma (see text). B: T1 sequence postgadolinium magnetic resonance image (MRI) in coronal plane of the patient in A. The expansile mass is again apparent with heterogeneous features centered along the mandibular ramus and involving the temporalis muscle into the suprazygomatic masticator space. |
arose from the laryngotracheal cartilages.16 While a rare site for chondrosarcoma, the cricoid lamina of the larynx is a unique tumor of generally indolent behavior.
TABLE 29.3 Principal Histologic Types of Bone Sarcoma Selected from the Second Edition (1993) World Health Organization (WHO) Classification of Bone Tumors | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
with Li-Fraumeni syndrome for the development of osteosarcoma, again with or without radiotherapy.24
and an accompanying soft tissue mass. In the end, it is our impression that MRI generally provides superior depiction of intraand extraosseous tumor in most malignant tumors of bone. Axial imaging complemented by either coronal or sagittal imaging planes using T1- and T2-weighted SPIN echo sequences most often provides accurate depiction of intra- and extraosseous tumor. To improve conspicuity, these sequences could be augmented by fat-suppressed pulse sequences. The maximum dimension of the tumor must be measured prior to any treatment.
 FIGURE 29-3. Flow schema for the assessment and management of soft tissue sarcoma (STS) of the head and neck. Because of the heterogeneity of tumor and anatomical sites, individualization is also needed. Thus some diseases, such as rhabdomyosarcoma, may need additional investigations (e.g., bone marrow examination)— see text for details of management. MRI, magnetic resonance imaging; CT, computed tomography; AJCC, American Joint Committee for Cancer; UICC, International Union Against Cancer; TNM, tumor, node, metastasis; RT, radiation therapy. |
of grade) and anatomic factors.32 However, a major limitation of the staging system is that it does not take into account the anatomic and histologic heterogeneity of these lesions. The system is also optimally designed to stage extremity tumors and is also applicable to the head and neck although lacks subtlety since the T-category size criterion dwarfs the anatomic sites of origin in the head and neck, which tend to be much smaller. An additional issue concerns rhabdomyosarcoma, where two separate descriptions of disease extent exist.
 FIGURE 29-4. A: Magnetic resonance image (MRI) used for fusion in the axial plane for computed tomography (CT) intensity-modulated radiotherapy (IMRT) planning of an unresectable malignant fibrous histiocytoma of the right base of skull. Shown are the 66.5 Gy isodose (95% of the prescribed dose of 70 Gy in 35 fractions) and the 56 Gy peripheral isodose line for radical IMRT for this lesion. MRI imaging shown on the right side is far superior to the CT planning data set on the left in delineating the lesion depicted as the gross tumor volume (GTV) in red in both images. B: The sagittal view of the radiotherapy plan shows the 66.5 Gy isodose (95% of the prescribed dose of 70 Gy in 35 fractions) and the 56 Gy peripheral isodose line for a course of radical radiotherapy alone in this patient. There is dose reduction to protect the optic chiasm using IMRT (arrow). C: The initial coronal MRI is shown with an enhancing mass centered along the right lateral wall of the sphenoid sinus and projecting medially into the sphenoid sinus and laterally into the middle cranial fossa. The lesion was considered unresectable because of involvement of the cavernous sinus with encasement of the right internal carotid artery. D: The patient experienced a complete response to fractionated photon irradiation using IMRT delivered stereotactically with 3-year follow-up and is currently asymptomatic without evidence of disease. |
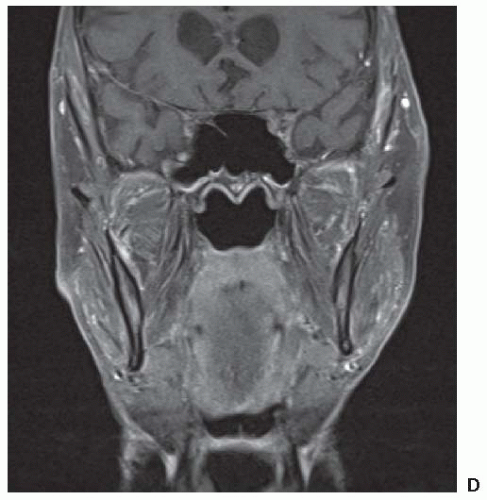 FIGURE 29-4. Continued |
of the classification used, adequate imaging of regional lymph nodes is imperative in patients in RMS.
TABLE 29.4 American Joint Committee on Cancer (AJCC) TNM Classification (7th Edition) of Soft Tissue Sarcomas | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
TABLE 29.5 Intergroup Rhabdomyosarcoma Study Group Postsurgical Grouping Classification | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||
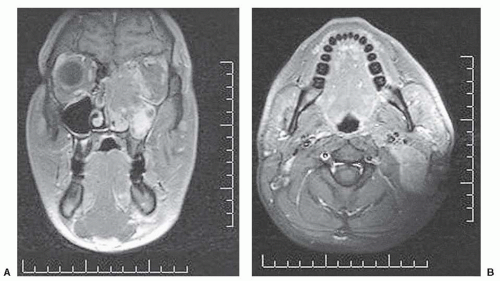 FIGURE 29-5. Magnetic resonance image of an alveolar rhabdomyosarcoma of the ethmoid sinus in a young adult. A: coronal view showing extensive orbital invasion; B: axial view showing concurrent extensive regional lymphadenopathy; C: complete response following combination chemotherapy with cyclophosphamide, actinomycin-D, vincristine (CAV), and etoposide with ifosfamide. Although consolidation radiotherapy commonly achieves local-regional control in these responsive tumors, such patients are at extremely high risk of failure in bone marrow, lung, and leptomeningeal sites. |
inability to deliver aggressive treatments because of their location in the critical anatomy of the head and neck. It may also relate to problems in delivering care for these rare diseases, if undertaken in the absence of a full multidisciplinary team. However, properly deployed principles and approaches should be able to achieve results in the head and neck that are comparable to similar lesions in extremity sites. Several randomized controlled trials have collectively established important milestones in the evolution of the local management of STS. With one exception, these trials have focused on extremity lesions and around the themes of surgery and adjuvant radiotherapy. These results are also highly relevant to the head and neck because of the similarity of biologic behavior of STS across body sites, even if the different histologic subtypes are not equally distributed by site.
 FIGURE 29-5. Continued |
TABLE 29.6 International Society of Pediatric Oncology (SIOP) Presurgical Staging Classification (Clinical and Radiologic Staging) | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
TABLE 29.7 American Joint Committee on Cancer (AJCC) TNM Classification (7th Edition) of Bone Sarcomas | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
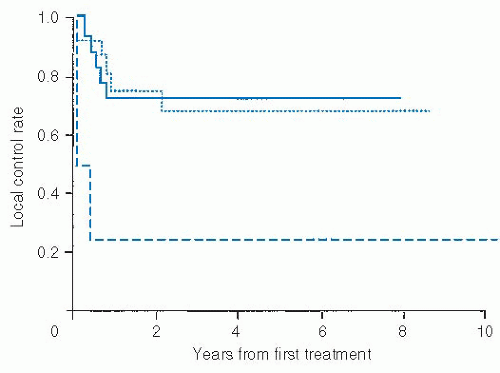 FIGURE 29-6. Actuarial estimate of local control rate in soft tissue sarcoma of the head and neck by surgical margins. Histopathologically clear (solid line), microscopically positive (dotted line), and gross disease (dashed) line are shown for patients treated with curative intent following surgical resection. Source: Data reproduced with permission from Le Vay J, O’Sullivan B, Catton C, et al. An assessment of prognostic factors in soft-tissue sarcoma of the head and neck. Arch Otolaryngol Head Neck Surg. 1994;120:981-986. |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


