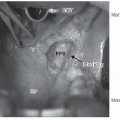
the paraganglia. The carotid body is a discrete oval structure situated behind the carotid bifurcation and receives blood supply directly from the carotid bifurcation via the glomic arteries. The afferent reflex is mediated by a branch of the glossopharyngeal nerve (nerve of Hering) (Fig. 27-2).7
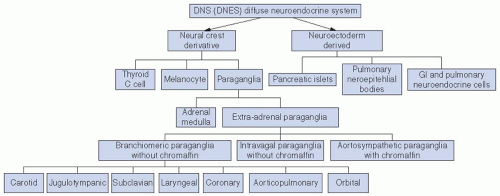 FIGURE 27-1. Classification of the diffuse neuroendocrine system. GI, gastrointestinal. |
patients with a paraganglioma. Since multicentric tumors may be metachronous, routine follow-up MRI, 111indium pentetreotide (OctreoscanR) or (18)F-DOPA positron emission tomography imaging is indicated.
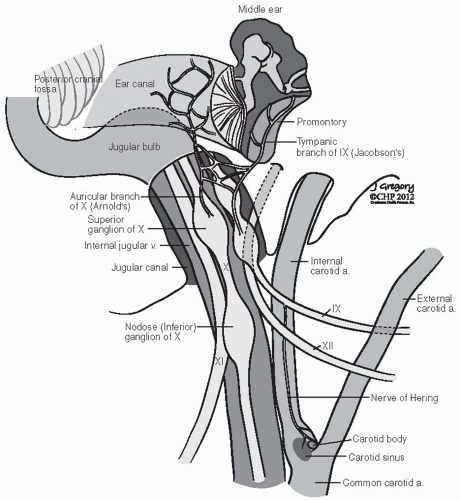 FIGURE 27-2. Schematic representation of lower cranial nerves at the level of jugular foramen. (See color insert.) |
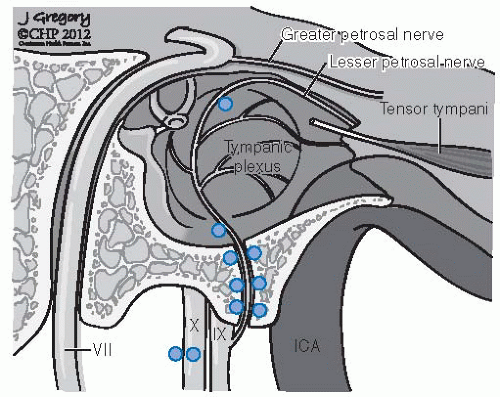 FIGURE 27-3. Distribution of frequent locations for glomus bodies in the temporal bone. (See color insert.) |
 FIGURE 27-4. Biochemical pathway for catecholamine metabolism. DOPA, dopa dihydroxyphenylalanine. |
predisposition to head and neck paragangliomas and adrenal/extra-adrenal pheochromocytomas.41 This inherited tumorigenic predisposition is transmitted in an autosomal dominant fashion with age-dependent and incomplete penetrance. However, for SDHD located on chromosome 11q, a parent-of-origin effect is revealed as the disease is manifest almost exclusively when the mutation is transmitted from the father. A maternal imprinting has therefore been postulated, but despite the pattern of inheritance, SDHD shows bi-allelic expression in normal tissues and neural crest-derived tissues.20,31
Patients with paraganglioma develop tumors at a younger age than sporadic cases
In PGL1, PGL2, and PGL3, the genetic transmission is autosomal dominant, with highly variable expressivity and reduced penetrance. Genomic imprinting is seen in PGL1: the
paternally transmitted genes lead to tumor development and the maternally transmitted gene gives carrier status without developing tumors
PGL1 and PGL4 show multicentricity and pheochromocytomas
PGL1 has a high degree of penetrance whereas PGL4 shows moderate penetrance
PGL4 shows a marked increase in malignant paragangliomas
PGL3 present exclusively as benign paragangliomas with no multifocality and has no association with pheochromocytomas
Other tumor syndromes: Neurofibromatosis type 1, MEN type 2, and Von Hippel-Lindau predispose to paragangliomas
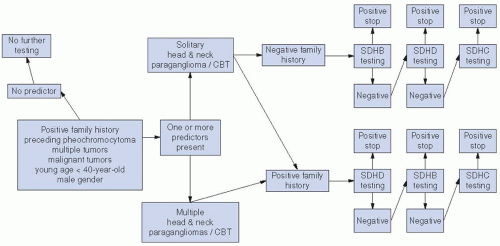 FIGURE 27-5. Algorithm for genetic testing for patients with head and neck paraganglioma. CBT, carotid body tumor. |
TABLE 27.1 Genetics of Paraganglioma: SDH- and SDH-related Genes | ||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
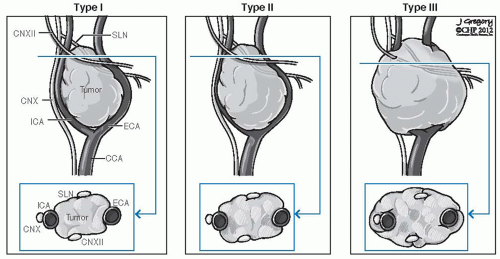 FIGURE 27-6. Shamblin’s classification of carotid body tumors. CCA, common carotid artery; CN, cranial nerve; ECA, external carotid artery; ICA, internal carotid artery; SLN, sentinel lymph node. |
TABLE 27.2 Shamblin’s Surgical Classification of Carotid Body Tumors | ||||||
|---|---|---|---|---|---|---|
|
Growth through Jacobson nerve canal and the hypotympanic air cells leads to involvement of the middle ear and mastoid bone leading to conductive hearing loss, tinnitus, and facial paralysis. Intracranial involvement through the jugular foramen is frequent. Erosion of the jugulocarotid spine leads to involvement of the vertical petrous carotid artery, and further anterior extension leads to involvement of its horizontal portion. More medial extension to the infralabyrinthine air cells lead to involvement of the petrous apex. Further medial extension along this pathway leads to involvement of the clivus and cavernous sinus. Occasionally, jugular paragangliomas will escape the confines of the temporal bone and involve the infratemporal fossa and parapharyngeal space. Lower cranial nerve involvement is frequent and ranges from 38% to 58%.58,59 Multiple cranial nerves are frequently involved in Vernet syndrome (paralysis of CNs IX, X, and XI) or Collet-Sicard syndrome (paralysis of CNs IX, X, XI, and XII). In at least 10% of jugular paragangliomas, one of these sequences is present (Fig. 27-7). The attendant symptomatology is hoarseness, swallowing difficulty, hemipalatal dysfunction with nasal air escape, shoulder motion restriction, and dysarthria due to tongue hemiparalysis. Two surgical classification systems are in wide use today. Both can be used preoperatively based on radiographic findings. The Fisch classification system makes no distinction between tympanic and jugular paragangliomas and is predicated on detailed patterns of progressive disease extension, whereas the Glasscock-Jackson system treats tympanic and jugular paragangliomas differently27,58 (Tables 27.3 and 27.4).
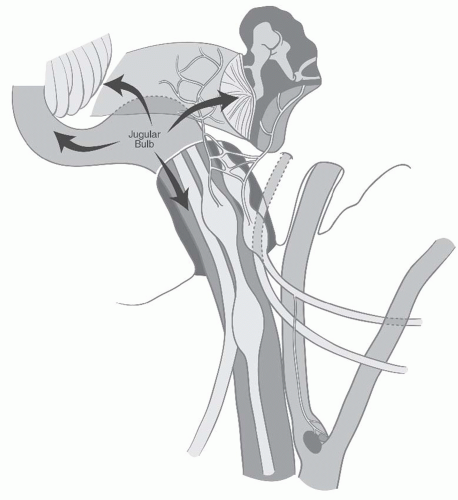 FIGURE 27-7. Modes of spread for jugular paragangliomas. |
transformation. Attention should be focused on a familial history of paragangliomas, MEN type 2, and Von Hippel-Linday syndromes. Genetic testing for PGL gene should be done if there is a positive family history, and other members of the family should be screened for paragangliomas.
TABLE 27.3 Fisch Classification of Jugulotympanic Paragangliomas | ||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||
TABLE 27.4 Glasscock-Jackson Classification of Jugulotympanic Paragangliomas | ||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||
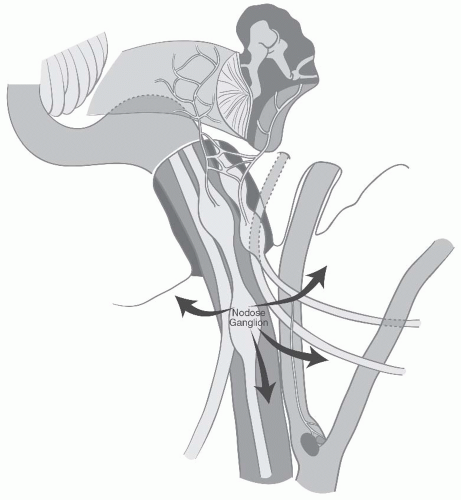 FIGURE 27-8. Modes of spread for vagal paragangliomas. |
 FIGURE 27-9. Computed tomography angiogram of a patient with a large paraganglioma. A: Volumetric surface rendering shows anterolateral displacement of the internal carotid artery and narrowing of its lumen. Note the filling of the internal jugular vein with contrast posterior to the common carotid artery, demonstrating the high flow within this tumor. B: Coronal reformat demonstrates involvement of the jugular foramen as well as the hypoglossal canal by tumor. C: Sagittal reformat demonstrates circumferential involvement of the internal carotid artery with anterior displacement, with extensive involvement of the jugular foramen to its intracranial portion. (See color insert for part A.) |
 FIGURE 27-10. Jugular paraganglioma. A: Coronal enhanced fat-suppressed T1-weighted image shows avid enhancement with focal round flow voids indicating large feeding vessels. B: Axial fat-suppressed T2-weighted image demonstrates the proximity of the tumor to the vertical portion of the petrous internal carotid artery as well as the intracranial but extradural component of the tumor in the jugular foramen posteriorly. |
 FIGURE 27-11. Vagal paraganglioma. Sagittal (A) and coronal (B) enhanced fat-suppressed T1-weighted images demonstrate typical rostrocaudal growth. The internal carotid artery is displaced anteriorly. |
that involve the ICA because of its vulnerability to injury during surgery, or where a planned internal carotid resection is contemplated. An angiographic balloon occlusion test involves the use of a femoral-artery-introduced catheter guided ultimately into the internal carotid, which is temporarily occluded usually at the carotid siphon within the cavernous sinus. This helps to determine whether there will be neurologic deficit.17,19 During occlusion, several methods of neurologic monitoring testing can be utilized. In order of decreasing sensitivity, they are clinical neurologic examination, electroencephalography (EEG), and quantitative blood-flow examination through xenon CT concurrent examination. Xenon CT angiography is a precise quantitative study with the best sensitivity, but is rarely available and cumbersome (Fig. 27-12).68,69
 FIGURE 27-12. Balloon occlusion testing. A: Frontal angiographic image shows the inflated radiodense balloon in the proximal left internal carotid artery, occluding flow. B: Quantitative cerebral blood flow images created by inhaling xenon gas during dynamic CT. The color images reveal reduced flow (blue instead of red) in the left MCA distribution. |
medical comorbidities, the type of paraganglioma in question (vagal, tympanic, jugular, carotid body), the presence of concurrent paragangliomas, the extent of the tumor, as well as any preoperative cranial nerve deficits, whether partial or complete. When surgery is contemplated, the availability of a surgical team with experience in lateral skull base methods, as well as an experienced interventional neuroradiology team, is sought.
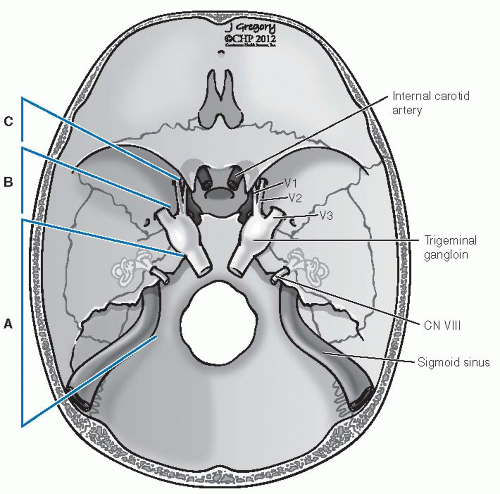 FIGURE 27-13. Fisch classification of infratemporal fossa approaches. Type A, exposure to the level of the anterior Eustachian tube, middle meningeal artery/foramen spinosum. Type B, exposure to the foramen ovale, V3. Type C, exposure to the foramen rotundum, V2. CN, cranial nerve. |
is the escape of embolizing particles into the cerebral circulation with all risk of stroke. This can occur through external-internal carotid circulation anastomoses or flow reversal from the arterial supply of the tumor. The advantages of embolization are tumor shrinkage and decreased blood flow that in large tumors have profound benefits. Less intraoperative bleeding is generally associated with a much easier dissection—more obvious tissue planes and less risk to normal anatomic structures, especially cranial nerves. Importantly, such a circumstance lessens the likelihood of requiring transfusion. Larger paragangliomas have multiple arterial feeding vessels that require superselective angiography. Using these methods, each successive embolization devascularizes additional compartments of the tumor until an absence of a tumor “blush” is achieved. It is essential that surgery be performed within 48 hours of embolization to avoid recruitment of collateral circulation. The inflammatory tumor reaction that has been reported following embolization is countered somewhat by the administration of steroids (Fig. 27-15).72,76
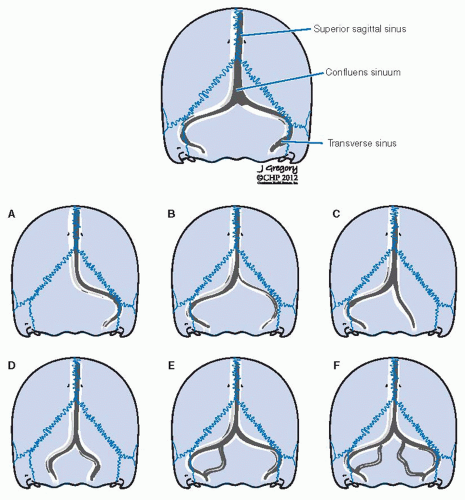 FIGURE 27-14. Variations of the intracranial posterior venous outflow system at the level of the transverse sinus, sigmoid sinus, and jugular bulb. Variations A and B would have a high surgical significance if a lesion were to involve the dominant side. |
tumors usually show marked hyperemia of the vasa nervosum of the sheath. In larger tumors, these nerves—vagus, hypoglossal, and glossopharyngeal—may be intimately involved and their dissection can cause dysfunction.
 FIGURE 27-15. Preoperative embolization of a carotid body tumor. A: Preembolization lateral angiogram shows early, intense blush reflecting high vascular flow with multiple collateral feeding vessels. B: Postembolization frontal angiogram shows marked reduction in vascularity. The primary feeding vessel (arrow) terminates abruptly. |
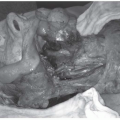
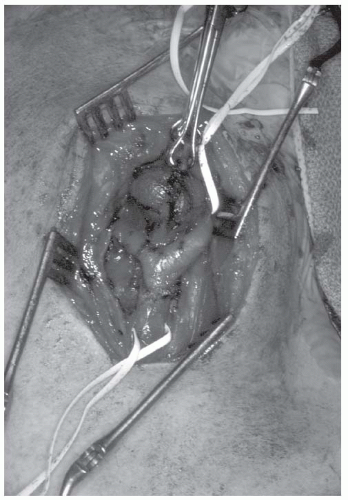 FIGURE 27-16. Intraoperative picture of a carotid body tumor. Tenaculum is on the lateral tumor surface. Vessel loops are around the common carotid artery (inferiorly) and internal carotid artery (superiorly). |
caution since the cranial nerve rootlets of the glossopharyngeal and the vagus are at their most vulnerable. In jugulotympanic paragangliomas, these rootlets are displaced medially and are thus in a favorable position. If the tumor does not extend into the pars nervosa/medial jugular bulb compartment, preservation of the cranial nerves is desirable.78
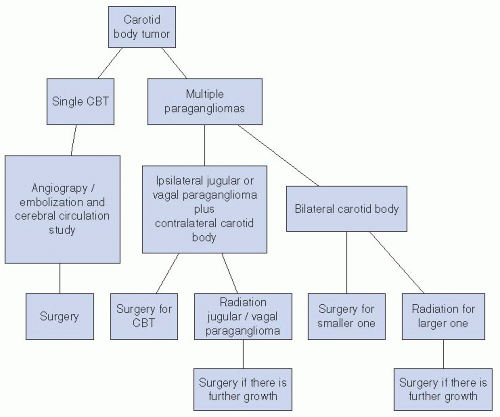 FIGURE 27-17. Decision analysis tree for the treatment of carotid body tumors (CBTs). |
nerve deficits are to be expected as these tumors extend medial to the cranial nerve rootlets, within the pars nervosa of the jugular foramen.
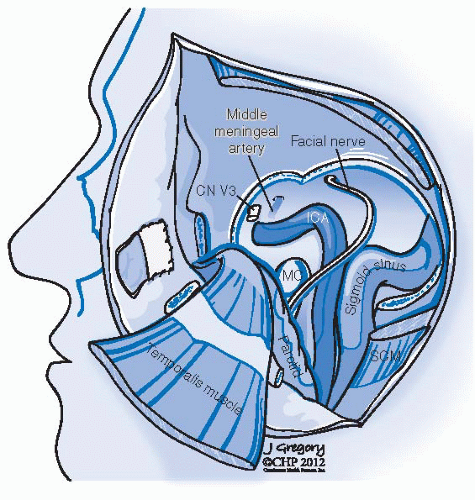 FIGURE 27-18. Schematic representation of a type B infratemporal fossa approach. Lateral temporal bone has been removed exposing the petrous carotid artery, jugular vein, jugular bulb and sigmoid sinus. Facial nerve has been mobilized in its horizontal and vertical portions. The third division of the trigeminal nerve (V3) has been divided as well as the middle meningeal artery right behind it. CN, cranial nerve; MC, mandibular condyle; SCM, sternocleidomastoid muscle; ICA, internal carotid artery. |
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


