Fish Oils in the Treatment of Dyslipidemia and Cardiovascular Disease
Harold E. Bays
Elevated triglycerides (TG) are a risk factor for cardiovascular disease (CVD). Severe hypertriglyceridemia can cause pancreatitis. Fish oils rich in omega-3 fatty acids lower TG and may have other cardiovascular and noncardiovascular benefits. Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are examples of omega-3 fatty acids, which are polyunsaturated fatty acids found in the oil of fish and other seafood. The American Heart Association (AHA) recommends fish/seafood consumption or fish oil supplementation as a therapeutic strategy to decrease cardiac dysrhythmias, reduce sudden death, decrease the rate of atherosclerosis, and to slightly lower blood pressure. In addition to fish/seafood consumption and fish oil supplements, a prescription omega-3 fatty acid (POM3) formulation is also available. In the United States, POM3 has regulatory approval at 4 g/day for treatment of patients with very high (> 500 mg/dL) TG. In Europe, POM3 is approved at 2 to 4 g/day to lower TG and at 1 g/day for secondary prevention of sudden death in those with CVD.
To achieve the often recommended combined EPA and DHA intake of about 1 g/day, an approximately fourfold or greater increase in the intake in these two omega-3 fatty acids than currently consumed in the United States would be required. Omega-3 fatty acids may have numerous cardiovascular and noncardiovascular health benefits that warrant such recommendations; but the main lipid effect of omega-3 fatty acids is to reduce TG. Hypertriglyceridemia is a common dyslipidemia in patients, who may or may not have other concurrent lipid abnormalities such as elevated low-density lipoprotein cholesterol (LDL-C) or decreased high-density lipoprotein cholesterol (HDL-C).
According to the National Health and Nutrition Examination Surveys (NHANES), mean LDL-C declined from 138 mg/dL in 1976-1980, to 123 mg/dL in 1999-2002 (1). This downward trend was most likely due to increased use of cholesterol-lowering drugs (such as statins), although some dietary changes in the population (such as lower dietary intake of total fat, saturated fat, and cholesterol) may have contributed as well. In contrast, in the same NHANES analysis, TG modestly increased, going from a mean of 114 mg/dL in 1976-1980, to 122 mg/dL in 1999-2002. This increase in TG is likely related, at least in part, to the substantial increase in obesity during these same time periods and occurred despite any increased use of lipid-altering drugs or beneficial changes in dietary fat intake. The results from NHANES 1999 to 2002 indicated that 25% of women and 35% of men had TG of 150 mg/dL or higher, a threshold beyond which is considered “normal.”
Specifically, the National Cholesterol Education Program, Adult Treatment Panel III (NCEP ATP III) defines “normal” TG as <150 mg/dL, “borderline high” TG as 150 to 199 mg/dL, “high” TG as 200 to 499 mg/dL, and “very high” TG as 500 mg/dL or more. TG that exceed 150 mg/dL is one component of the “metabolic syndrome”(2), a constellation of multiple CVD risk factors that tend to cluster together. Other components include low HDL-C (<40 mg/dL in men; <50 mg/dL in women), high blood pressure (≥130/≥85 mm Hg), elevated blood glucose (>100 mg/dL), and increased waist circumference (>40 inches in men; >35 inches in women).
Given the inadequate omega-3 fatty acid intake in countries such as the United States, the increasing prevalence of hypertriglyceridemia, the contribution of elevated TG to CVD risk, and the potential cardiovascular benefits of omega-3 fatty acids, organizations such as the AHA recommend fish oil intake, with the amount being based upon CVD risk (3). For patients without known CVD, the AHA recommends eating a variety of (preferably fatty) fish at least twice a week, as well as oils and foods rich in α-linolenic acid (ALA) (flaxseed, canola, and soybean oils; flaxseed and walnuts). For patients with CVD, the AHA recommended consuming about 1 g of EPA and DHA per day, preferably from fatty fish, with administration of encapsulated EPA and DHA formulations as an alternative approach. Finally, for hypertriglyceridemic patients in need of therapy for elevated TG, the AHA recommends 2 to 4 g of EPA and DHA per day provided as capsules.
THEORETICAL CONSIDERATIONS
Biochemistry
Fats, Omega-3 Fatty Acids, Lipid Transport, and Free Fatty Acid Release from Adipose Tissue
The potential mechanisms of action of omega-3 fatty acids are best understood within a framework of lipid metabolism, lipoprotein-mediated lipid transport, and free fatty acid (FFA) release from adipose tissue (4,5,6).
Types and Source of Different Fatty Acids. Foods quantitatively differ in the amount of fat content and qualitatively differ in the fatty acid components of TG. Saturated fatty acids contain no double bonds between their carbon atoms since all of the available bonds are occupied with hydrogen atoms. Monounsaturated fatty acids contain one double bond. Polyunsaturated fatty acids have more than one double bond between their carbon atoms. The predominant fatty acid in TG determines whether it is referred to as a saturated fat, monounsaturated fat, or polyunsaturated fat. Many animal
and dairy products are high in saturated fats. Examples of foods high in monounsaturated fats include olive oil and canola oil. Corn oil and safflower oils are rich in polyunsaturated fats. Fish and other seafood contain variable amounts of all of three types of fatty acids, with some fish having high concentrations of omega-3 polyunsaturated fatty acids.
and dairy products are high in saturated fats. Examples of foods high in monounsaturated fats include olive oil and canola oil. Corn oil and safflower oils are rich in polyunsaturated fats. Fish and other seafood contain variable amounts of all of three types of fatty acids, with some fish having high concentrations of omega-3 polyunsaturated fatty acids.
Transport and Fate of Exogenous TG. Blood lipids depend upon endogenous synthesis and dietary consumption. Thus, the type and amount of dietary fats is medically relevant because dietary fat consumption contributes to the lipid content of body organs, such as adipose tissue. Over 90% of consumed fats are in the form of TG, which are emulsified by bile acids into micelles, and then hydrolyzed in the intestine by intestinal and pancreatic lipases. The di- and monoglycerides, glycerol, and FFA derived from digested fats then enter intestinal cells, where FFA may become re-esterified into TG, and then packaged along with re-esterified cholesterol into apoB48-containing chylomicrons (see Chapter 7). Chylomicrons are then secreted into mesenteric lymph vessels and eventually into the circulation for delivery of lipids to peripheral tissues. Body fat represents the major storage of lipids and energy in the body, with the greatest amount of fat being located in adipose tissue. Other important fat stores are located in liver and muscle. TG represent the major molecular constituent of stored fat and are composed of a three-carbon glycerol backbone with a fatty acid being attached to each carbon (see also Chapter 7).
Essential Fatty Acids. ALA is a common plant polyunsaturated omega-3 fatty acid that contains 18 carbons and three double bonds. Linoleic acid (LA) is also a plant polyunsaturated fatty acid that contains 18 carbons, but in contrast to ALA has two double bonds, the first of which is six carbons from the omega end, and thus an example of an “omega-6 fatty acid.” EPA and DHA are examples of omega-3 polyunsaturated fatty acids found in fish oils and other seafood. EPA has 20 carbons and five double bonds (“eicosa” = 20, “penta” = 5; thus “eicosapentaenoic” acid). DHA has 22 carbons and six double bonds (“docosa” = 22, “hexa” = 6; thus “docosahexaenoic” acid) (Fig. 21.1). Both ALA and LA are termed “essential” fatty acids because they are not synthesized in humans and must be ingested. Humans do have enzymes (called desaturases) that convert ALA into EPA and subsequently EPA into DHA. However, the activities of these enzymes are limited. Thus, EPA and DHA are often described as also being “essential,” because in humans, the only way to achieve optimal omega-3 fatty acid tissue levels is through dietary consumption. In other words, dietary intake is not only necessary for supplying “essential” fatty acids but may also be important in achieving desired concentrations of other favorable fatty acids that have a low potential for intrinsic synthesis, such as EPA and DHA.
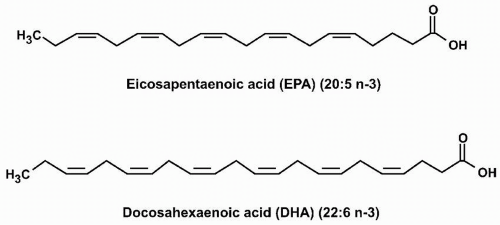 FIGURE 21.1 Eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA) are omega-3 fatty acids with five and six double bonds, respectively, with the first double bond three carbons from the methyl or “omega” end. |
Free Fatty Acids. While most fatty acids are transported esterified to TG carried on lipoproteins, some fatty acids circulate “free,” that is, unesterified and bound to albumin, from which they may be delivered to adipocytes, muscle, or liver. Short-chain FFA (<6 carbons), especially those containing two to four carbons, are more water soluble, owing to their polar carboxyl group. As the number of carbons increases progressively to medium-chain FFA (6 to 12 carbons) and long-chain FFA (12 carbons or more), the FFA become more nonpolar and less water soluble. Quantitatively, the amount of FFA in the body is much less compared to the amount of esterified fatty acids carried by lipoproteins or stored in adipose tissue. However, if adiposity results in adiposopathy wherein adipose tissue is unable to adequately store FFA, and if this results in an increase in circulating long-chain, saturated FFA, then an increased delivery of these kinds of FFA to peripheral tissues may result in “lipotoxicity”, which is clinically manifested by insulin resistance in muscle and liver (7), decreased insulin secretion from the pancreas (8), and an increased risk of sudden cardiac death (9). Conversely, increased circulating and tissue omega-3 fatty acids reduce the risk of sudden cardiac death, illustrating how omega-3 fatty acids have fundamentally different clinical effects compared to other fatty acids.
Because it is the major source of fasting, circulating FFA, adipose tissue plays a major role in FFA metabolism, and the types and amount of FFA released into the circulation from adipose tissue are dependent upon several key factors including dietary intake (see also above), the prandial state (i.e., after meals, increased insulin secretion markedly reduces circulating FFA), and an underlying pathophysiologic environment (e.g., insulin resistance increases circulating FFA). In insulin resistance, insulin does not adequately inhibit hormone-sensitive lipase, leading to a net increase in release of FFA. Physical exercise may transiently increase FFA release from adipose tissue to muscle for energy, as the result of lipolysis. However,
with prolonged physical exercise, fasting FFA may be decreased due to increased uptake by muscle and improved insulin sensitivity.
with prolonged physical exercise, fasting FFA may be decreased due to increased uptake by muscle and improved insulin sensitivity.
Adiposopathy. The relative “healthy” or “sick” state of adipose tissue itself has a profound effect on fatty acid metabolism. At least from a metabolic standpoint, the pathogenic potential of adipose tissue is more related to its dysfunction than to its mass (10). Adiposopathy, or “pathos” of adipose tissue, is anatomically manifested by adipocyte hypertrophy, visceral adiposity, growth of adipose tissue beyond its vascular supply, and ectopic fat deposition (10,11). If subcutaneous adipocytes fail to adequately proliferate or differentiate (i.e., adipogenesis), then they may become dysfunctional (“sick”) with a decreased capacity to store fats, resulting in a net increase in circulating FFA. An increased accumulation of visceral adipose tissue (a fat depot more enzymatically active than subcutaneous adipose tissue) may increase the delivery of FFA to the liver through the portal circulation. An increase in FFA released from adipose tissue may increase the risk of metabolic diseases such as type 2 diabetes mellitus, hypertension, and dyslipidemia (10).
Thus, depending upon extrinsic (metabolic environment) and intrinsic (inherent adipose tissue function) factors, FFA may undergo a net storage or net release from adipose tissue. The types of fatty acids stored in, and released from, adipose tissue are dependent upon enzymatic production within adipocytes, as well as the kinds of fatty acids originating from dietary consumption. Regarding storage, adipose tissue biopsy has sometimes substituted for frequent dietary analysis over a long period of time to determine fatty acid intake (such as omega-3 fatty acids), because fat cell fatty acid concentrations reflect dietary consumption and tissue concentrations (12). Regarding release, fatty acid mobilization from fat cells appears to be dependent upon the length of the fatty acid chains and degree of unsaturation. The types of fatty acids most commonly found in adipose tissue as components of TG include those with 12 to 24 carbon atoms and zero to six double bonds. The fatty acids that appear to be the easiest mobilized through hydrolysis by hormone-sensitive lipase are those in which the carbon chains are shorter and more unsaturated (13). For example, fatty acids that are 16 to 20 carbon atoms in length with two or more double bonds may be more easily mobilized than those with 20 to 24 carbon atom fatty acids with zero to one double bonds, which may be more weakly mobilized. In regard to omega-3 fatty acids, EPA (with 20 carbons) appears more readily released from adipose tissue than DHA (with 22 carbons) (14).
Metabolism
Metabolism of Fatty Acids in Liver, Lipoproteins, and Peripheral Tissues
Much of the efficacy of omega-3 fatty acids in reducing TG is thought to be due to their metabolic effects in the liver. In this way, omega-3 fatty acids differ from many other fatty acids. For example, omega-3 fatty acids decrease the synthesis of hepatic TG. In contrast, once many non-omega-3 fatty acids are delivered to liver, they may be re-esterified onto glycerol, producing TG. Increased hepatic TG are then associated with apoB-100 through the action of microsomal TG transfer protein, and the synthesis and secretion of very low density lipoprotein (VLDL) particles into the circulation is increased (see also Chapter 7). VLDL particles transport the majority of TG in the fasted state. Similar to chylomicrons, the TG on VLDL are hydrolyzed by lipoprotein lipase (LPL) and apolipoprotein C-II (apoC-II) into FFA and monoglyceride. The FFA can be taken up by the adipocytes and then re-esterified into TG that are stored for future energy use. Alternatively, FFA can be taken up by muscle cells where they are used for immediate energy needs, or FFA can be returned back to the liver. During hydrolysis of VLDL particles, intermediate-density lipoproteins (IDL) (VLDL remnants) are produced, which can be removed at the surface of the liver by the interaction of apoE with the LDL receptor (LDLR), or converted by hepatic lipase into cholesteryl ester-rich LDL (see also Chapters 1 and 8). Hydrolysis and removal of TG from VLDL also results in the eventual creation of LDL particles. These effects of many non-omega-3 fatty acids upon hepatic and circulatory lipid metabolism are very different than the lipid effects of omega-3 fatty acids.
Omega-3 Fatty Acid Lipid-Altering Mechanisms of Action. Although not completely understood, it is generally believed that omega-3 fatty acids reduce TG levels by decreasing hepatic VLDL synthesis and by increasing VLDL catabolism (5) (Fig. 21.2). Regarding synthesis, whether originating from chylomicrons, VLDL, or adipose tissue, if the delivery of many FFA increases hepatic fatty acid levels, then this increases the substrate for TG synthesis and VLDL secretion. However, an increase in the delivery of omega-3 fatty acids to the liver may impair hepatic production of fatty acids, by decreasing the enzymatic conversion of acetyl CoA into FFA (Fig. 21.2). They may also increase the β-oxidation of existing hepatic fatty acids and further decrease the substrate for TG synthesis. Additionally, omega-3 fatty acids may inhibit both phosphatidic acid phosphatase/phosphohydrolase (PAP), an enzyme that catalyzes the conversion of phosphatidic acid (PA) into diacylglycerol (DAG), and diacylglycerol acyltransferase (DGAT), an enzyme that catalyzes the final step in the synthesis of TG (Fig. 21.2). Another mechanism by which omega-3 fatty acids may reduce hepatic VLDL production is through increased degradation of apoB-100. Through these mechanisms, omega-3 fatty acids reduce VLDL production and thus decrease the levels of plasma TG.
Another manner in which omega-3 fatty acids may also reduce TG is through an increase in LPL activity. LPL metabolizes both VLDL and chylomicron particles into smaller and denser remnant particles, which become more depleted in TG, and more relatively enriched in cholesteryl esters. LPL also increases the conversion of VLDL into LDL. The increase in LPL activity with omega-3 fatty acids, and the effect upon TG-enriched lipoproteins, helps explain some of the seemingly paradoxical lipid effects found with their clinical use, for example, the rise in LDL-C with the decrease in VLDL.
Comparison of Mechanism of Action of Omega-3 Fatty Acids and Fibric Acid Derivatives. Fibric acid derivatives are agonists for peroxisome proliferator activating receptors-alpha (PPAR), which are nuclear receptors that induce the LPL gene and suppress the inhibitor of LPL, apolipoprotein C-III (apoC-III), leading to significant reductions in TG (see Chapter 23). In hypercholesterolemic patients with low or borderline TG,
fibrates may significantly lower LDL-C. If baseline TG levels are high (˜400 mg/dL) at baseline in patients without hypercholesterolemia, fibrates may reduce TG by 50%, but mildly increase (not decrease) LDL-C levels by ˜3%. With even greater TG elevations (>700 mg/dL), LDL-C levels may be even more increased to as much as 50%. This can largely be attributable to increased LPL activity, which converts TG-rich lipoproteins (e.g., as VLDL and IDL) into LDL particles (see also Chapter 21). Given that LDL-C levels are the primary treatment target to reduce CVD risk, such rises in LDL-C with therapies such as fibrates may prompt the use of combination therapy with agents that predominantly reduce LDL-C (see Chapter 23).
fibrates may significantly lower LDL-C. If baseline TG levels are high (˜400 mg/dL) at baseline in patients without hypercholesterolemia, fibrates may reduce TG by 50%, but mildly increase (not decrease) LDL-C levels by ˜3%. With even greater TG elevations (>700 mg/dL), LDL-C levels may be even more increased to as much as 50%. This can largely be attributable to increased LPL activity, which converts TG-rich lipoproteins (e.g., as VLDL and IDL) into LDL particles (see also Chapter 21). Given that LDL-C levels are the primary treatment target to reduce CVD risk, such rises in LDL-C with therapies such as fibrates may prompt the use of combination therapy with agents that predominantly reduce LDL-C (see Chapter 23).
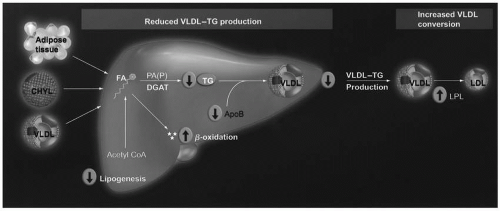 FIGURE 21.2 Potential TG-lowering mechanisms of eicosapentaenoic acid and docosahexaenoic acid. Pathogenic adipose tissue, increased postprandial CHYL, and increased VLDL particles may normally increase free FA delivery to the liver, and increase hepatic lipid content, which are substrates for TG synthesis and, thus, increase VLDL production. In contrast, most evidence supports that omega-3 fatty acids inhibit hepatic TG synthesis, decrease VLDL production/secretion, and increase VLDL metabolism by decreasing lipogenesis by decreasing the enzymatic conversion of acetyl CoA into FAs; increasing β-oxidation of FA; inhibiting both PAP (an enzyme that catalyzes that reaction of converting PA into DAG) and DGAT (an enzyme that catalyzes the final step in TG synthesis); potentially increasing the degradation of apolipoprotein B; and increasing LPL activity, which is an enzyme that increases the conversion of VLDL particles into LDL particles. CHYL, chylomicrons; DAG, diacylglycerol; DGAT, diacylglycerol acyltransferase; FA, fatty acid; LPL, lipoprotein lipase; PA, phosphatidic acid; PAP, phosphatidic acid phosphatase/phosphohydrolase; TG, triglyceride; VLDL, very low density lipoprotein. (From Harris WS, Bulchandani D. Why do omega-3 fatty acids lower serum triglycerides? Curr Opin Lipidol. 2006;17: 387-393.) (see color insert.) |
As is true with fibrates, omega-3 fatty acids also lower TG and not uncommonly increase LDL-C in hypertriglyceridemic patients. The total number of LDL particles appears to remains relatively constant with a decrease in the small LDL particles and an increase in large LDL particles (Fig. 21.3). No clinical trial data exist that this rise in LDL-C represents harm or potential “toxicity” to patients. In fact, most evidence supports that omega-3 fatty acids reduce cardiovascular risk as do the fibrates. Importantly, clinical trials mostly support that even with increases in LDL-C, omega-3 fatty acids decrease the total cholesterol (TC) carried by atherogenic lipoproteins, as reflected by decreased non-HDL-C levels (TC minus HDL-C). This is primarily due to a reduction of TG, VLDL, and their remnants. If omega-3 fatty acids do reduce atherosclerosis, then it is likely the lowering of non-HDL-C that probably best explains this favorable effect. This may be of clinical significance given that non-HDL-C may be a better predictor of CVD risk than LDL-C alone.
Pathophysiology of Hypertriglyceridemia
Metabolic Justification for Therapeutic Intervention with Omega-3 Fatty Acids
Omega-3 fatty acids not only lower TG but also may improve clinical abnormalities often found in hypertriglyceridemic patients (5).
Pancreatitis. The clinical importance of elevated TG is multifold. In particular, patients with very high TG (≥500 mg/dL) are at increased risk for pancreatitis. VLDL particles normally constitute approximately 90% of the TG-containing lipoproteins, particularly in the fasting state. However, while both VLDL and chylomicron-associated TG increase after meals, pancreatitis most often occurs due to profound postprandial increase in chylomicrons in patients with an underlying genetic defect exacerbated by some secondary cause of dyslipidemia (see Chapter 7). Chylomicrons and their remnants may impair pancreatic capillary blood flow, causing ischemia-induced disruption in acinar structure, and the necrosis, edema, and inflammation characteristic of pancreatitis. TG-induced pancreatitis most often occurs with TG levels >1,000 mg/dL. However, TG levels vary considerably from day to day and throughout the day. Furthermore, when fasting TG levels are
500 mg/dL or higher, postprandial TG levels may be much higher. Therefore, the NCEP ATP III has recommended that fasting TG levels ≥500 mg/dL be treated to prevent pancreatitis, which is a disease process with significant potential morbidity, and in severe cases, substantial risk of mortality. Omega-3 fatty acid therapy can lower very high TG, and although not specifically evaluated in patients with pancreatitis, POM3 is indicated for treatment of TG ≥500 mg/dL that may lead to pancreatitis.
500 mg/dL or higher, postprandial TG levels may be much higher. Therefore, the NCEP ATP III has recommended that fasting TG levels ≥500 mg/dL be treated to prevent pancreatitis, which is a disease process with significant potential morbidity, and in severe cases, substantial risk of mortality. Omega-3 fatty acid therapy can lower very high TG, and although not specifically evaluated in patients with pancreatitis, POM3 is indicated for treatment of TG ≥500 mg/dL that may lead to pancreatitis.
 FIGURE 21.3 Revealing the underlying atherogenic potential of hypertriglyceridemia. Many patients with hypertriglyceridemia have increased cholesterol carried by atherogenic particles, which is best assessed by measuring non-HDL-C levels. VLDL particles are considered to be atherogenic. Omega-3 fatty acid therapy decreases the cholesterol carried by VLDL particles and is a cholesterol effect not typically measured in clinical practice. Omega-3 fatty acids may also decrease VLDL particle size. Conversely, omega-3 fatty acids may increase LDL-C levels, which is a lipid parameter that is often measured in clinical practice. This is thought to be due to the increased conversion of VLDL particles into LDL particles. Finally, omega-3 fatty acids may increase LDL particle size, which may render them less atherogenic. Overall, despite a potential increase in LDL-C levels, many studies have reported that POM3 reduces non-HDL-C, which may be a better predictor of atherosclerotic coronary heart disease risk than LDL-C alone. HDL-C, high-density lipoprotein cholesterol; LDL-C, low-density lipoprotein cholesterol; POM3, prescription omega-3-acid ethyl esters; VLDL, very low density lipoprotein. (From Bays HE, Tighe AP, Sadovsky R, et al. Prescription omega-3 fatty acids and their lipid effects: physiologic mechanisms of action and clinical implications. Expert Rev Cardiovasc Ther. 2008;6:391-409.) |
CVD. Hypertriglyceridemia is also a risk factor for CVD; however, it may not always be an independent CVD risk factor. This is important to keep in mind in making therapeutic decisions regarding the most appropriate initial lipid-altering therapy administered for the purpose of reducing CVD risk—including the use of omega-3 fatty acids. In fact, depending upon the underlying lipoprotein abnormality, even the most severe cases of hypertriglyceridemia may not be a risk factor for increased CVD risk (4,6). This is in stark contrast to the clinical consequences of severe hypercholesterolemia, as occurs in patients with heterozygous and particularly homozygous familial hypercholesterolemia. Severely hypercholesterolemic patients clearly have a marked increased risk of CVD. Furthermore, CVD outcomes trials have demonstrated that reducing elevated LDL-C reduces the risk of CVD events—an effect that has yet to be definitively shown by reducing TG per se. For these reasons, LDL-C remains the primary lipid treatment target for the purpose of reducing CVD risk (see also Chapters 5 and 7).
Demonstrating an independent association of elevated TG and CVD risk is challenging because many other metabolic abnormalities often accompany hypertriglyceridemia, such as hyperglycemia, obesity, high blood pressure, low HDL-C, elevated apoB-100, and increased numbers of small dense LDL particles (see Chapter 8). Despite the lack of a prospective
CVD outcome trial involving TG as the primary end point, the totality of evidence supports elevated TG as being atherogenic, depending upon the cause of the elevated TG and associated metabolic abnormalities. While it is entirely indicated to initiate drug therapy first to achieve LDL-C treatment goals and reduce CVD risk (see Chapters 5 and 23), omega-3 fatty acids may often help improve a number of remaining metabolic abnormalities associated with increased CVD risk in patients with persistently elevated TG.
CVD outcome trial involving TG as the primary end point, the totality of evidence supports elevated TG as being atherogenic, depending upon the cause of the elevated TG and associated metabolic abnormalities. While it is entirely indicated to initiate drug therapy first to achieve LDL-C treatment goals and reduce CVD risk (see Chapters 5 and 23), omega-3 fatty acids may often help improve a number of remaining metabolic abnormalities associated with increased CVD risk in patients with persistently elevated TG.
Related posts:
 Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
 Dyslipidemia in Children and Adolescents
Dyslipidemia in Children and Adolescents
 Dietary Treatment of Dyslipidemia
Dietary Treatment of Dyslipidemia
 Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
 Dyslipidemia and Atherosclerosis in Women
Dietary Treatment of Dyslipidemia
Dyslipidemia and Atherosclerosis in Women
Dietary Treatment of Dyslipidemia
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


