Factor V and Factor VIII
Randal J. Kaufman
Philip J. Fay
Laura Popolo
Thomas L. Ortel
Factor V and factor VIII are two homologous nonenzymatic proteins that play a critical role in blood coagulation, an essential process that involves the sequential activation of plasma proteases in response to blood vessel injury. Factor V and factor VIII circulate in plasma in inactive forms that are devoid of coagulant activity. In their active state, factors Va and VIIIa serve as cofactors for their respective proteases (factor Xa and factor IXa), resulting in a striking enhancement of their catalytic activity.
To maximally express their function, factor VIIIa and factor Va need to assemble into enzyme complexes. Factor VIIIa together with factor IXa, calcium ions, and phospholipid membranes forms the intrinsic Xase complex. This complex catalyzes the proteolytic conversion of the serine protease factor X to factor Xa. Subsequently, factor Xa, factor Va, Ca2+, and phospholipid membranes form the prothrombinase complex that converts prothrombin to thrombin. The thrombin-mediated conversion of soluble fibrinogen to insoluble fibrin generates a stable platelet-fibrin clot at the site of vascular injury. Thus, factor V and factor VIII catalyze pivotal steps in the coagulation cascade. This is also underscored by the clinical findings, which indicate that conditions of factor V or factor VIII deficiency lead to parahemophilia and hemophilia A, respectively.
The activation of factor V and factor VIII consists of proteolytic cleavage catalyzed by thrombin or factor Xa. The activation process has been the subject of intense investigation during the last two decades. Since proteolysis is an irreversible protein modification, it needs to be spatially and temporally controlled. Efficient inactivation mechanisms targeting the cofactors exist and are essential for normal hemostasis. One key reaction in down-regulating coagulation is the inactivation of factor Va by the anticoagulant-activated protein C (APC). Cleavage of factor Va at specific sites converts it into factor Vai with a complete loss of cofactor activity. Factor VIIIa is also a substrate of APC, but the intrinsic instability of factor VIIIa may make this reaction irrelevant from a biologic perspective.
Although the knowledge of factor V and factor VIII and their regulation has progressed significantly over the last two decades, the molecular mechanisms by which protein cofactors factor V and factor VIII accelerate substrate cleavage remains to be fully defined. The difficulties in having homogeneous preparations of these factors were partially overcome by the advent of recombinant DNA technologies. However, the dynamics and interactions of these factors with partners (ions, membranes, activators and inhibitors, plasma proteins) are many, and their analyses require a reductionist approach and a rather complex process of critical integration and interpretation of the results obtained. The molecular models that were recently built on the basis of the crystal structures of factor VIII, factor Vai, and their interacting proteins (proteases and substrate-proteases) provide a background to gain insight into the mechanistic details of how the structure of these factors changes to fulfill their function.
FROM GENE TO PROTEIN
Factor V and Factor VIII: Historical Background
Coagulation factor V and factor VIII were discovered in the 1930s and 1940s. Quick1 first reported the identification of a labile factor in plasma that was necessary for the rapid conversion of prothrombin to thrombin. At approximately the same time, Owren2 identified a patient with a severe bleeding disorder (parahemophilia), who lacked this factor. This protein, which was the fifth coagulation factor to be discovered, was designated factor V. Subsequent work established that activated factor V functions as a nonenzymatic cofactor in the prothrombinase complex.3 The observation that a factor V mutation is the most common risk factor for venous thrombosis4 has drawn attention to this protein, leading to many advances in the field.
Factor VIII identification was tightly linked to characterization of hemophilia A, a severe bleeding disorder caused by deficiency of factor VIII. Transfusion of whole blood was shown to successfully treat a hemophilia-associated bleeding episode by 1840.5 In 1911, Addis6 demonstrated that normal plasma can shorten the clotting time of hemophilic blood. It was 25 years later when Patek and Taylor described the role of factor VIII in hemostasis and designated the factor as “antihemophilic globulin.”7 In the 1950s, plasma concentrates and then in 1965, cryoprecipitates were developed for treatment of bleeding episodes associated with hemophilia A. In the early 1980s, factor VIII was purified from human plasma8 and subsequently the gene was cloned. At approximately the same time, the human factor V gene was isolated. Over the past three decades, considerable efforts have been made defining the cofactor activity and its regulation.
Organization of Factor V and Factor VIII Genes
The genes encoding human factor V and factor VIII,9,10 named F5 and F8, respectively, have an almost identical exon-intron organization.11,12 F5 is localized to human chromosome 1q21- 25.13 The gene spans approximately 80 kilobase (kb) pairs of DNA and consists of 25 exons and 24 introns (FIGURE 12.1). The exons range in size from 72 to 286 base pairs (bp) with the exception of exon 13, which spans 2,820 bp. The introns range in size from 448 bp to 11.4 kb. The length of F5 mRNA is approximately 7 kb.14,15,16 At the present time, there is no evidence for alternative splicing of F5 transcripts, and the mechanisms
responsible for the control of transcription and translation remain unknown. A rich report of this gene and the surrounding genomic region is available at NCBI (http://www.ncbi.nlm. nih.gov/gene/2153).
responsible for the control of transcription and translation remain unknown. A rich report of this gene and the surrounding genomic region is available at NCBI (http://www.ncbi.nlm. nih.gov/gene/2153).
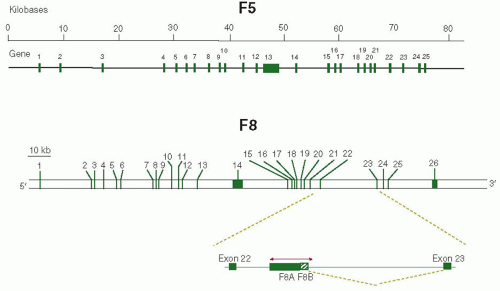 FIGURE 12.1 Features of the factor V and factor VIII encoding genes. The top line shows the scale of F5 genomic sequence. The second line shows the intron-exon organization of F5 with the exons being represented by green boxes. For F8, the bar represents the scale, and the bottom of the figure shows the intron-exon organization with exons represented by green boxes. The genomic region between exons 22 and 23 is enlarged to show the direction of transcription of F8A and F8B (see text). |
F8 is localized on human X chromosome (Xq28). The gene is 186 kb long (˜0.1% of the DNA of the X chromosome) and contains 26 exons and 25 introns (FIGURE 12.1). The exon length varies from 69 to 262 bp except for exon 14 that is 3,106 bp and the last exon 26 that has 1,958 bp. There are some large intervening sequences including IVS22 that is 32 kb long. F8 mRNA is approximately 9 kb, of which the coding sequence covers 7,053 nucleotides. In the gene, a CpG island within IVS22 is associated with two additional transcripts. One transcript of 1.8 kb is produced abundantly in a wide variety of cells. The orientation of this transcript is opposite to that of factor VIII and contains no intervening sequence.17 This cDNA of 1.7 kb, termed factor VIII-associated gene A (F8A), is conserved in the mouse.18 The second transcript of 2.5 kb, named factor VIII-associated gene B (F8B), is transcribed in the same direction as factor VIII, and after a short exon that may encode for eight amino acids, it utilizes exons 23 to 26 of the F8 gene19 (FIGURE 12.1). The two transcripts F8A and F8B originate within 122 bases from each other. The sequences of F8A and F8B along with a few kilobases of surrounding DNA are also present in two other areas of the X chromosome approximately 400 kb telomeric to F8.17,20 The function of these transcripts and their potential protein products is unknown. Transgenic mice with a deletion of the F8B gene showed particular eye abnormalities, suggesting that migration of neural crest cells might have been perturbed during eye development.21 For a detailed report and graphical view of F8 and its genomic context, see http://www.ncbi.nlm.nih.gov/gene/2157.
Features of Factor V and Factor VIII Proteins
F5 and F8 encode large precursor proteins of 2,224 and 2,351 amino acids, respectively. At the N-terminal, both precursors have a signal peptide, of 19 and 28 amino acids, respectively, that is cleaved upon translocation in the lumen of the endoplasmic reticulum (ER). Factors V and VIII share a common organization of three structural domains that occur in the same order A1-A2-B-A3-C1-C2 (FIGURES 12.2 and 12.3) and a high degree of amino acid identity (about 40%) over the entire length except for the B domains where there is no similarity. The B domain is encoded by an unusually large single exon spanning 3.1 and 2.8 kb in FVIII and FV, respectively. In general, the B-domain sequence is about 900 amino acids in length and rich in flexible amino acids (e.g., serine, threonine, glycine, and proline) and N-and O-glycosylation sites. A unique feature of both B domains is their unusual number of N-linked oligosaccharides. The factor VIII and factor V B-domains have 18 and 25 N-linked glycosylation sites, respectively. In the human factor V B-domain, two tandem repeats of a 17-amino acid sequence and 31 tandem repeats of a nine-amino acid sequence are present (FIGURE 12.2). The consensus sequences for these two types of repeats are SQDTGSPSXMPRPWEDXP and [T,N,P]LSPDLSQT. The significance of the tandem repeats structures that are present in the B domain of factor V, but not factor VIII, remains to be determined.
The three A-type domains present in factor V and factor VIII are approximately 330 amino acids in length, and are
about 30% identical to the triplicated A-type domains in the copper-binding proteins ceruloplasmin and hephaestin and to the single A domain of ferroxidase.9,10,16,22 The C-type domains in factor V and factor VIII are approximately 150 amino acids in length and are homologous with discoidin 1, a lectin that mediates cell adhesion and migration in the single-cell organism Dictyostelium discoideum. Discoidin 1 domains are found in a growing family of proteins implicated in cell adhesion and/or developmental processes.23,24
about 30% identical to the triplicated A-type domains in the copper-binding proteins ceruloplasmin and hephaestin and to the single A domain of ferroxidase.9,10,16,22 The C-type domains in factor V and factor VIII are approximately 150 amino acids in length and are homologous with discoidin 1, a lectin that mediates cell adhesion and migration in the single-cell organism Dictyostelium discoideum. Discoidin 1 domains are found in a growing family of proteins implicated in cell adhesion and/or developmental processes.23,24
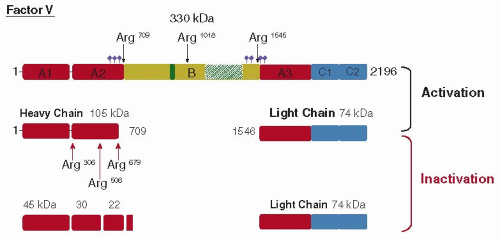 FIGURE 12.2 Domain structure and proteolytic activation/inactivation of human factor V. The structural domains of the single-chain factor V are depicted in the top line by the labeled boxes: (i) a triplicated A domain of approximately 330 amino acids; (ii) a unique B domain of 890 amino acids; and (iii) a duplicated C domain of approximately 150 amino acids. The tandem repeats of 2 × 17 amino acids are represented by the green box and the 9 × 31 amino acids by the dashed green box. The location of potential disulfide bonds and asparagine-linked glycosylation sites is not shown for simplicity (see Accession number P12259 at UniprotKB: http://www.uniprot.org/uniprot/). The violet diamonds indicate the location of putative sulfotyrosines. The thrombin cleavage sites are indicated by the black arrows at the top of the figure. The location of the APC cleavage sites are indicated by red arrows at the bottom of the scheme. The apparent molecular mass of the intermediates and final products following activation by thrombin and inactivation by APC are indicated. The heavy chain and light chain of thrombin-activated factor V (Va) form a calcium-dependent heterodimer. The numbering refers to the amino acid sequence devoid of the signal sequence. |
In addition to these domains, in factor VIII there are three acidic amino acid-rich regions (a1, a2, a3) that reside at the junction of the A1/A2 (a1: residues 336 to 372), A2/B (a2: residues 711 to 740), and B/A3 (a3: residues 1649 to 1689) domains (FIGURE 12.3). Moreover, both factor V and factor VIII have been shown to bind copper and calcium ions.25,26
Within the secretory pathway, factor VIII is processed to a heterodimer of the heavy-chain fragment of variable size (apparent molecular weight ˜90 to 200 kDa)7,8 in a metal-dependent association with the light chain of 80 kDa (FIGURE 12.3). The A1, A2, and B domains occur in the heavy chain whereas the A3, C1, and C2 domains occur in the light chain. In plasma, human factor VIII is present at a concentration of approximately 1 nM (150 ng/mL) and is stabilized by association with a 50-fold molar subunit excess of von Willebrand factor (vWF). Human factor V circulates in the plasma as single-chain glycoprotein of high apparent molecular weight (˜330 kDa) (FIGURE 12.2). Its concentration is approximately 30 nM (10 µg/mL).
The analysis of homologies of the primary sequences of factors V and factors VIII from different species suggests that these genes evolved from an ancestral ferroxidase gene by triplication of the A domain, insertion of the B domain, and addition of the C domains to the primordial cofactor gene. Subsequently, the factor V and factor VIII genes possibly evolved by duplication and divergence of amino acid residues within the B domain, although the high number of N-glycosylation sites were preserved, while amino acid sequences within the A and C domains were conserved.
Biosynthesis of Factor V and Factor VIII
Factor V Expression
The main site of factor V biosynthesis is the liver. Human megakaryocytes also have been shown to contain factor V mRNA and to express factor V27 but it is believed that most or all of platelet factor V originates from the plasma pool by endocytosis.28,29 As uptake of factor V by megakaryocytes has not been demonstrated, the origin of platelet factor V in humans remains controversial.30 However, total factor V is distributed into two pools: 20% is contained in platelet granules whereas the remaining 80% is in the plasma compartment. The storage of factor V within platelets appears to be important because, when platelets aggregate to form the primary hemostatic plug, they also provide both a surface for assembly of the prothrombinase complex as well as high local concentrations of released platelet factor V. In addition, residual platelet factor V supports thrombin generation in patients with severe factor V deficiency and mild bleeding symptoms.31 The synthesis of factor V by several other cell types, including bovine aortic endothelial cells, bovine vascular smooth muscle cells, and human mesangial cells, has also been reported, although the physiologic importance of synthesis at these sites remains unknown.
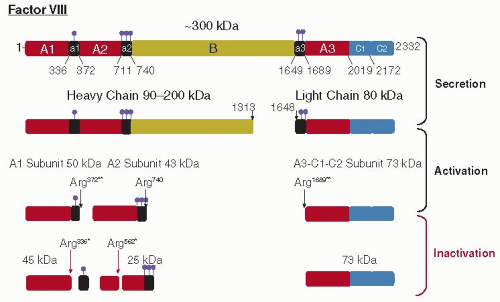 FIGURE 12.3 Domain organization and proteolytic activation/inactivation of human factor VIII. The structural domains of the full-length factor VIII are depicted in the top line by the labeled boxes: (i) a triplicated A domain of about 330 amino acids, (ii) a unique B domain of 980 amino acids, and (iii) a duplicated C domain of 150 amino acids. The black boxes represent regions enriched in acidic amino acids. The ball-stick symbol (violet) represents the positions of sulfotyrosine residues that were experimentally determined (residues 346, 718, 719, 723, 1664, 1680). The location of free cysteine residues, disulfide bonds, and asparagine-linked glycosylation sites is not shown (see Accession number P00451 at UniprotKB Database: http://www.uniprot.org/uniprot/). Within the Golgi compartment in the cell, factor VIII is cleaved at two sites within the B domain to generate heavy-chain polypeptides of variable size (90 to 200 kDa) and the 80-kDa light chain. The cleavage sites for activation by thrombin (black arrows) and inactivation by APC (red arrows) are shown at the point of the processed products. The apparent molecular mass (kDa) of the intermediates and final products are indicated. The two cleavages required for thrombin activation (**) and the sites of proteolytic inactivation by APC (*) are indicated. The numbering refers to the amino acid sequence devoid of the signal sequence. |
Factor VIII Expression
Although the natural cell type that produces factor VIII has not been definitively identified, liver transplantation studies in factor VIII-deficient dogs32 and several hemophiliac patients33 strongly implicate the liver and/or the reticuloendothelial system as primary sites of factor VIII synthesis. Initial immunochemical localization by light microscopic34 or electron microscopic35 examination detected the factor VIII antigen in hepatocytes. However, RNA hybridization analysis detected factor VIII mRNA in hepatocytes as well as in many other cells and tissues.36 More recent analysis of factor VIII mRNA by quantitative reverse transcription polymerase chain reaction identified highest levels in liver, followed by kidney.37 Factor VIII mRNA and protein were concentrated in hepatic sinusoidal endothelial or Kupffer cells, with significantly less in hepatocytes.37 In addition, isolated sinusoidal endothelial cells were shown to produce factor VIII.38 Also a part of factor VIII is contained in a granules of platelets together with factor V.39 Since there are no known natural established cell lines that express factor VIII, it has not been possible to study the biosynthesis of factor VIII in its native host cell.
Trafficking and Modification within the Cell
Factor V and factor VIII are abundantly modified glycoproteins secreted into the blood. The factor VIII biosynthetic pathway was proposed based on analysis of factor VIII expression in transfected cell lines, such as Chinese hamster ovary cells (FIGURE 12.4). On synthesis, factor VIII is translocated into the lumen of the ER where the signal peptide is cleaved and the protein is subjected to N-linked core glycosylation (FIGURE 12.4 steps 1 to 3, see also Chapter 5 “Cell Biology, Protein Processing, and Cell Signaling”). A significant portion of newly synthesized factor VIII is retained in the ER through interactions with two protein chaperone systems designed to prevent the exit of unfolded proteins from the ER. First, factor VIII is bound to BiP, the most abundant ER protein40 (FIGURE 12.4, step 4). BiP expression is induced by glucose deprivation, inhibition of N-linked glycosylation, or the presence of malfolded protein within the ER. In addition, high-level factor VIII expression can also induce transcription of the BiP gene indicating the induction of the unfolded protein response (for details, see Chapter 5).41 The level of BiP in the cell inversely correlates with the efficiency of factor VIII secretion.42,43 BiP exhibits a peptide-dependent adenosine triphosphatase activity. Factor VIII dissociation from BiP and secretion requires an unusually high level of intracellular adenosine triphosphate (ATP).44 Because factor VIII interacts with BiP, and factor V does not, it was possible to identify a primary BiP-binding site by analysis of the secretion of factor VIII and factor V chimeric proteins. This study identified a hydrophobic beta-sheet within the factor VIII A1 domain that resides adjacent to a proposed ligand, Cys310, for binding copper ion that was also recently confirmed by the crystal structure of FVIII (see further FIGURE 12.6).26,45,46 Exposed hydrophobic residues are
preferred sites of BiP binding. Substitution of phenylalanine at residue 309 with serine within the hydrophobic sheet increased the secretion efficiency of factor VIII several-fold and this correlated with a reduced requirement for ATP for secretion.45 These studies suggest that BiP may have a role in copper ion binding to factor VIII. Finally, a portion of factor VIII forms high molecular weight aggregates immediately after synthesis.47 These aggregates require ATP for disassembly and secretion.47
preferred sites of BiP binding. Substitution of phenylalanine at residue 309 with serine within the hydrophobic sheet increased the secretion efficiency of factor VIII several-fold and this correlated with a reduced requirement for ATP for secretion.45 These studies suggest that BiP may have a role in copper ion binding to factor VIII. Finally, a portion of factor VIII forms high molecular weight aggregates immediately after synthesis.47 These aggregates require ATP for disassembly and secretion.47
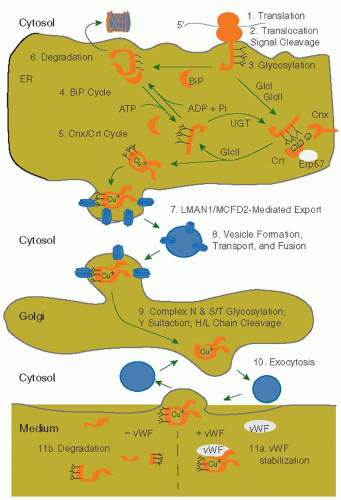 FIGURE 12.4 Synthesis, processing, and secretion of factor VIII in mammalian cells. The figure depicts the pathway of biosynthesis of factor VIII from the primary translation product that is translocated into the lumen of the ER to the medium as determined by expression in culture cells. For detail in the description see text under “Trafficking and modification within the cell.” |
The second chaperone system that retains factor VIII in the ER recognizes the structure on N-linked oligosaccharides on factor VIII. Rapid trimming of the two outermost glucose residues of the N-linked glycans (N-acetylglucosamine2-mannose9-glucose3) prepares protein-bound N-glycans for association with the two homologous ER lectins calnexin (CNX) and calreticulin (CRT) and exposure to the glycoprotein-specific oxidoreductase ERp57 to catalyze proper disulfide bond rearrangement (see Chapter 5).48,49 Release from CNX/CRT depends on glucosidase II cleavage of the innermost glucose residue. Although the modalities of misfolded protein domain recognition and ER retention are not known,50,51 the enzyme UDPglucose: glycoprotein glucosyltransferase (UGT) and the CNX/CRT chaperone system are thought to play central roles.52,53 Current models52,53,54,55,56,57 propose that UGT reglucosylates non-native polypeptides thereby preventing their release from the CNX/CRT chaperone system and their exit from the ER. Factor VIII interacts with both CNX and CRT, although their roles in factor trafficking within the ER are not known.58
Formation of disulfide bonds by oxidation of a pair of cysteine residues is another crucial modification for proteins destined to be secreted and contributes to the stabilization of the structure. Factor VIII contains eight disulfide bonds and factor V has seven putative disulfide bonds deduced by similarity. The enzyme PDI1 plays the predominant function as catalyst of oxidative folding in the ER. Interestingly, it has been recently reported that treatment with antioxidants improves secretion of factor VIII from cultured cells suggesting that folding of complex multidomain proteins can generate oxidative stress.59
Improperly folded proteins are selectively extracted from the ER and directed for degradation by the cytosolic proteasome in a process called ER-associated degradation (ERAD, step 6 in FIGURE 12.4). A significant portion of factor VIII within the ER never transits to the Golgi compartment, but rather is
degraded through ERAD (see Chapter 5). Factor VIII within the ER is retrotranslocated and degraded by the cytosolic 26S proteasomal machinery. Recently, ER degradation enhancing mannosidase-like (EDEMs) proteins were shown to recognize oligosaccharide structures trimmed by ER mannosidase I and mediate their transfer to the cytosol for ERAD.60
degraded through ERAD (see Chapter 5). Factor VIII within the ER is retrotranslocated and degraded by the cytosolic 26S proteasomal machinery. Recently, ER degradation enhancing mannosidase-like (EDEMs) proteins were shown to recognize oligosaccharide structures trimmed by ER mannosidase I and mediate their transfer to the cytosol for ERAD.60
Finally, studies on combined deficiency of factors V and VIII indicate that oligosaccharides are also important for trafficking of factor VIII and V from the ER to the Golgi compartment through interaction with the cargo receptor complex LMAN1/MCFD2 (FIGURE 12.4, step 7). The sum of these findings supports the notion that oligosaccharide processing plays a central role in directing factor VIII trafficking within the secretory pathway.
The portion of factor VIII that is secretion competent transits to the Golgi apparatus, where the majority of factor VIII is processed at two sites within the B domain after residues 1313 and 1648 to generate the heavy chains (90 to 200 kDa) and the light chain (80 kDa) (FIGURES 12.3 and 12.4). Also within the Golgi apparatus, factor VIII is further processed by (a) modification of the asparagine-linked high mannosecontaining oligosaccharides to complex types, (b) addition of carbohydrate to multiple serine and threonine residues within the B domain (O-glycosylation), and (c) addition of sulfate to six tyrosine residues within the heavy and the light chains61 (FIGURE 12.4, step 9).
Factor VIII is finally secreted by exocytosis (FIGURE 12.4, step 10). In the plasma, vWF stabilizes factor VIII through noncovalent association. In vitro studies demonstrate that vWF can alter intracellular transport and secretion of factor VIII from the cell. vWF promotes the association of the light and heavy chains of factor VIII upon secretion from the cell and results in stable accumulation of factor VIII activity in the conditioned medium of factor VIII-producing cultured mammalian cells62,63 (FIGURE 12.4, step 11a). In the absence of vWF in the conditioned medium, the factor VIII heavy and light chains are secreted into the medium as dissociated chains that are subsequently degraded (FIGURE 12.4, step 11b).
In vitro reconstitution experiments demonstrated that vWF can directly promote reassembly of isolated heavy and light chains of factor VIII,62,64 suggesting a possible role of vWF in facilitating factor VIII assembly. The effect of vWF in promoting factor VIII heavy and light chain assembly and stable secretion in cell culture systems may reflect the role of vWF in regulating levels of factor VIII activity in vivo.65,66,67,68 However, it is not known whether vWF also regulates factor VIII protein synthesis and/or secretion in vivo.69 At present, there are no natural cell types that are known to express both factor VIII and vWF.70 It is proposed that a portion of factor VIII synthesized is coexpressed with vWF from an endogenous endothelial bed and secreted as a complex upon stimulation,71 although this has not been directly demonstrated in vivo.
Metal Ions in Factor V and Factor VIII
Both factors V and VIII bind calcium and copper ions and iondependent interactions appear vital for both their structure and function. The recently reported three-dimensional (3-D) structures of these factors identified the position of the high-affinity ion-binding sites and provided new interpretations on the role of ions in factor V and VIII structure and function. Adams et al.72 solved the structure of bovine factor Vai (A1/A3-C1-C2) (FIGURE 12.5). The two A domains sit on a platform formed by the two C domains, which are aligned in an edge-to-edge fashion, a feature that was found to be present also in the crystal structure of a recombinant single-chain B-domain-deleted human factor VIII (FIGURE 12.6B) and in an intermediate-resolution crystallographic structure of a recombinant form of factor VIII consisting of a heterodimer of the A1-A2 and A3-C1-C2 subunits.73
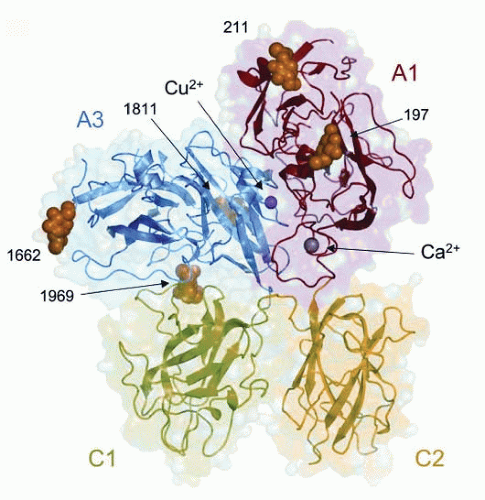 FIGURE 12.5 The 3-D structure of bovine factor Vai. Domain arrangement in a ribbon representation of bovine factor Vai, indicating the positions of the metal ions within the A1 and A3 domains (Ca2+: gray and Cu2+: pink) and of five N-linked chains (orange) at residues N197, N211, N1811, N1662, and N1969. The van der Waals representation in the background shows the surface contour of the protein. The C domains are side-by-side and predict a role in membrane binding (see text). (Adapted from Adams TE, et al. The crystal structure of activated protein C-inactivated bovine factor Va: implications for cofactor function. PNAS. 2004;101:8918-1923. Use with permission © 2004 by PNAS). |
A high-affinity calcium-binding site and a copper-binding site were identified in factor Vai. Chain association is required for factor V function and is dependent on a divalent cation. Although calcium ion was believed to bridge the heavy and light chains,74 the structure revealed that the high-affinity calcium-binding site is located entirely in the A1 domain74 (FIGURE 12.5). The side chains of both Asp111 and Asp112, along with the main-chain carboxyl oxygens of Lys93 and Glu108, coordinate the calcium ion. The structural studies clearly indicate that chain association cannot directly be attributed to the coordination of calcium but rather calcium may play a role in the maintenance of the A1-C3 domain interface.
The role of copper ion in factor V has always been uncertain. A single copper ion-binding site is located in the A3 domain of bovine factor Vai with ligands His1802, His1804, and Asp1844
arranged in a trigonal planar coordination geometry.72 Recently, Song et al.74 used recombinant factor V mutants to demonstrate that Asp111 in the heavy chain was important for the metal ion binding and for interactions between the heavy and light chains of the active cofactor. However, the structure of factor Vai suggested that copper ion provides additional stabilization of the A1-A3 interface rather than directly linking the two domains.
arranged in a trigonal planar coordination geometry.72 Recently, Song et al.74 used recombinant factor V mutants to demonstrate that Asp111 in the heavy chain was important for the metal ion binding and for interactions between the heavy and light chains of the active cofactor. However, the structure of factor Vai suggested that copper ion provides additional stabilization of the A1-A3 interface rather than directly linking the two domains.
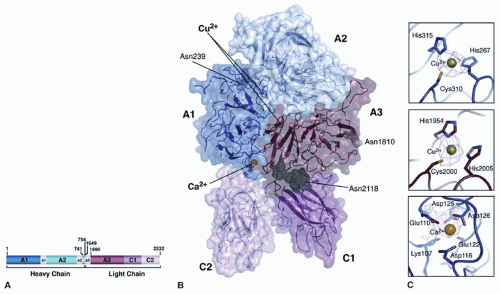 FIGURE 12.6 Three-dimensional structure of B-deleted domain factor VIII. (A) Domain organization of the B domain-deleted human factor VIII used for x-ray crystallography. (B) Structure of B-domain-deleted factor VIII. The A1 domain and a2 acidic regions are colored in dark blue. The A2 domain and the a2 acidic region are in light blue. The A3 domain, the C1 domain, and the C2 domain are colored dark red, dark pink, and light pink, respectively. The a3 acidic region is disordered and not included in the structure. Two Cu2+ ions are located internally within the A1 and A3 domains (green), one Ca2+ (yellow) is bound within the A1 domain and three N-linked oligosaccharide chains (gray) are indicated by arrows. (C) The amino acids that define the ion-binding sites are shown in detail (for a description, see text). (Adapted from Ngo JC, et al. Crystal structure of human factor VIII: implications for the formation of the factor IXa-factor VIIIa complex. Structure 2008;16:597-606. Use with permission © 2008 by Elsevier Ltd). |
The crystal structures of factor VIII revealed one calcium-binding site and two copper-binding sites. The position of the calcium-binding site in the A1 domain confirms the region in the A1 domain identified in factor V as a calcium-binding site. In the structure of factor VIII, the calcium is liganded by the carboxyl groups of Glu110, Asp116, Asp125, and Asp126 and the backbone carbonyl of Glu122 and Lys107 as observed for factor V74 (FIGURE 12.6C). It is noteworthy that mutations of these residues (Asp125Ala and Asp126His) were associated with mild/moderate hemophilia A, whereas mutations at the remaining residues (Glu110Val, Asp116Tyr, Glu110Val, Lys107Thr, and Glu122Lys) were related with severe hemophilia A (http://hgdb. org.uk/). Recent mutational data further support the existence of this calcium-binding site in both factors Va and VIIIa.74 The calcium ion is unlikely to play a role in directly bridging the light and heavy chains but could be involved in the maintenance of the interface between C2 and A1 domains in a given conformation. In addition, the intermediate-resolution x-ray crystal structure predicts a second calcium-binding site within the A2 domain utilizing residues Asp538 and Asp542.73
While previous results indicated a role of calcium in promoting the active conformation of factor VIII, copper ion was primarily involved in enhancing intersubunit affinity.75 The two copper-binding sites are within the A1 and A3 domains (FIGURE 12.6B). In the A3 domain, the site is composed of His1954, Cys2000, and His2005 and is dispensable for cofactor function.76 However, a His1954Leu mutation associated with mild hemophilia A has been reported (http://hgdb.org.uk/). The second site in the A1 domain (composed of ligands His267-Cys310-His315) is essential for cofactor function (FIGURE 12.6C). Electron paramagnetic spectroscopy suggests that cupric ion (Cu+) occupies this site.26 The mutation of Cys310Ser destroyed factor VIII activity and yielded secreted heavy and light chains that were not associated, supporting a requirement for this copper ion-binding site in heavy and light chain association. Interestingly, mutations of Cys310Phe and His267Pro have been reported in patients with severe hemophilia A, suggesting the
importance of this second copper-binding site (http://hgdb.org. uk/). However, this second copper site in the A1 domain localizes near the interface of the A1 and A3 domains rather than at the interface suggesting that a role for copper in enhancing interchain affinity is indirect.
importance of this second copper-binding site (http://hgdb.org. uk/). However, this second copper site in the A1 domain localizes near the interface of the A1 and A3 domains rather than at the interface suggesting that a role for copper in enhancing interchain affinity is indirect.
Copper alone is insufficient to generate cofactor activity but in the presence of calcium does marginally increase the specific activity of factor VIII.64,77 Recombining purified factor VIII chains in buffers containing Ca2+ or Mn2+ yields active factor VIII.62,64 These ions do not contribute to the interchain affinity but rather promote cofactor activity by modulating the conformation of the heterodimer.75
FUNCTION AND REGULATION OF FACTOR V AND FACTOR VIII
Cofactor Activities of Factor VIII and Factor V
Factor VIII acts to increase the rate of conversion of factor X to factor Xa by the protease factor IXa. The Km for factor X activation in the presence of factor IXa and calcium is decreased several orders of magnitude by phospholipid due to concentration of enzyme and substrate on the plane of the phospholipid surface (Table 12.1). Factor VIIIa dramatically increases the Vmax of factor X activation by factor IXa by 100,000-fold78 and consequently the catalytic efficiency (Table 12.1). Factor VIIIa accelerates the proteolysis of factor X by interaction with both factor X and factor IXa on a phospholipid surface to facilitate a conformational change in the active site of factor IXa that favors catalysis.79
In a similar manner, factor Va enhances the activation of prothrombin to thrombin by the serine proteinase factor Xa in the presence of calcium ions and a procoagulant phospholipid surface.3 As shown in Table 12.1, the acceleration of prothrombin activation by phospholipids results from a 1000-fold decrease in the Km for prothrombin.80,81 The major contribution of factor Va is a 3,000-fold increase in the Vmax for prothrombin activation80,81,82 (Table 12.1). The observed increase in Vmax is due, in part, to the fact that factor Va acts as a receptor for factor Xa resulting in an increased concentration of factor Xa at the membrane surface. Overall, the main effect of factor Va is to enhance the catalytic efficiency of prothrombin activation probably by properly orienting factor Xa and prothrombin.
Table 12.1 The effect of cofactors on the rate of activation of factor X and prothrombin | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Regulation of Factor VIII Cofactor Activity
Activation
The secreted heterodimer factor VIII is activated by limited proteolysis (for review, see Ref.83). Upon treatment of intact factor VIII with thrombin, there is a rapid >30-fold increase and subsequent first-order decay of procoagulant activity. Significant data has also characterized the activation of factor VIII by factor Xa, and other observations suggest that Factor XIa84 and VIIa85 can also catalyze this reaction. The physiologic activator(s) of factor VIII has (have) not been rigorously determined. Thrombin is the best characterized activator of factor VIII and maximal activation by thrombin is greater than that observed with factor Xa86.
Thrombin cleaves peptide bonds after Arg372, Arg740, and Arg1689. Procofactor activation coincides with proteolysis of both the heavy and light chains of factor VIII as depicted in FIGURE 12.3. Cleavage within the heavy chain at Arg740 generates a 90-kDa polypeptide that is subsequently cleaved at Arg372 to generate polypeptides of approximately 50 and 43 kDa.87 These polypeptides are designated as the A1 (residues 1 to 372) and A2 (residues 373 to 740) subunits, respectively, owing to their domainal derivations (FIGURE 12.3). Concomitantly, the 80-kDa light chain is cleaved at Arg1689 to generate a 73-kDa polypeptide88 designated as the A3-C1-C2 subunit (residues 1690 to 2332). The cleavage of selected bonds is likely to lead to the appropriate structural changes that impact cofactor function. Significant data support that thrombin anion-binding exosites I and II are involved in recognition of factor VIII.89,90,176,177 The corresponding binding sites on FVIII are thought to be the regions rich in acidic amino acids that contain the posttranslational modified amino acid tyrosine sulfate and border each of the cleavage sites91 (FIGURE 12.3). The tyrosine-sulfate residues appear to enhance thrombin cleavage at adjacent sites, but do not affect factor Xa cleavage.92 These observations suggest that thrombin may utilize tyrosine-sulfate residues to facilitate interaction and/or cleavage. In support of this contention are data showing that a mutation at a tyrosine sulfation site (Tyr346 to Cys) yielded a factor VIII that was defective in activation by thrombin93 and is associated to mild hemophilia A. However, conflicting data show that mutations of individual Tyr residues to Ala in the acidic region designated as a2 resulted in little, if any, effect on rates of thrombin-catalyzed cleavage or activation of factor VIII.94 Thus the contribution of sulfated Tyr residues to the activation mechanism may be restricted to selected regions in the procofactor.
Numerous studies have correlated the appearance of the 50-, 43-, and 73-kDa polypeptides with peak factor VIII activity.87,95,96 Other studies examined the role of the individual cleavage sites in the expression of FVIIa cofactor activity. Mutagenesis studies showed that cleavage after residue 740 was not required for cofactor activity.97,98 In contrast, mutation at either Arg372 or Arg1689
yielded molecules that were not cleaved by thrombin at the mutated site and were not susceptible to thrombin activation.97 The importance of cleavage at residues 372 and 1689 for factor VIII activation was demonstrated in vivo by the identification of hemophilia A patients having missense mutations that prevent cleavage at either Arg372 or Arg1689.99,100,101,102 These findings indicate that activation requires cleavage at both residues 372 and 1689, but does not appear to require a specific sequential order for cleavage at these sites. Cleavage at Arg1689 releases factor VIII from the inhibitory influence of vWF103 and appears to additionally increase the activity of factor VIIIa.104,105 Cleavage at Arg372 is essential to expose a functional factor IXa-interactive site(s) in the A2 domain that is cryptic in the unactivated molecule.106
yielded molecules that were not cleaved by thrombin at the mutated site and were not susceptible to thrombin activation.97 The importance of cleavage at residues 372 and 1689 for factor VIII activation was demonstrated in vivo by the identification of hemophilia A patients having missense mutations that prevent cleavage at either Arg372 or Arg1689.99,100,101,102 These findings indicate that activation requires cleavage at both residues 372 and 1689, but does not appear to require a specific sequential order for cleavage at these sites. Cleavage at Arg1689 releases factor VIII from the inhibitory influence of vWF103 and appears to additionally increase the activity of factor VIIIa.104,105 Cleavage at Arg372 is essential to expose a functional factor IXa-interactive site(s) in the A2 domain that is cryptic in the unactivated molecule.106
Examination of apparent rates of thrombin cleavage at the three sites in factor VIII shows that Arg740 > Arg1689 > Arg372. Thus, the step essential to forming cofactor activity, cleavage at Arg372 to liberate the A1 and A2 subunits, is rate limiting. Cleavage at Arg740 and Arg1689 appears to facilitate cleavage within the heavy chain as mutations at these sites slows subsequent cleavage at Arg372. However, the different magnitude of the effects produced by Arg1689Gln or Arg740Gln mutations on the reduction of the rate of cleavage of the light chain indicates that the docking of Arg740 represents the preferred pathway.107,108
Factor VIIIa is a heterotrimer of A1, A2, and A3-C1-C2 subunits105,109,110 (FIGURE 12.3). The A1 and A3-C1-C2 subunits retain the metal ion linkage and can be isolated as a stable A1/A3-C1-C2 dimer. Conversely, the A2 subunit is associated with the A1/A3-C1-C2 dimer in a primarily electrostatic interaction and readily dissociates from the dimer at physiologic pH and ionic strength (see below).
In an attempt to probe individual residues that contribute side chain hydrogen-bonding interactions to the association of A2 subunit in the cofactor, Wakabayashi et al. mutated 30 polar or charged residues to Ala at the borders of the A2 and A1, and A2 and A3 domains that were spatially separated by <2.8 Å.111,112 Results from that study showed that a number of the point mutations exhibited more rapid decay rates (5- to 40-fold) in factor VIIIa activity, hence showed reduced affinity for A2 subunit, suggesting a role for these residues in A2 subunit retention in the heterotrimer. Recent results have shown that replacing acidic residues such as E272, D519, E665, and E1984 with Ala or Val to yield hydrophobic interactions at the interfaces yielded factor VIII variants with as much as eightfold increases in the stability of factor VIIIa, as judged by reduced rates of activity decay.113 Furthermore, these mutations appeared to have an additive effect in that variants where more than one acidic residue was replaced with Ala or Val showed further increases in cofactor stability.112
Aside from identification of cleavage sites, the structural alterations that transform “procofactor” factor VIII into active cofactor, factor VIIIa, are not well characterized. Circular dichroism spectral analysis indicates that purified factor VIIIa shows significant reduction in random coil and increase in beta-sheet structures compared with factor VIII;114 however, it is unclear what contribution the B domain makes to these gross conformational differences. Zero-length cross-linking studies suggest formation of a new salt bridge between the N-terminal half of A2 subunit and the C-terminal region of A1 subunit following thrombin cleavage.115 Furthermore, new hydrogen-bonding interactions involving A2 domain appear to form following conversion of the procofactor to the active cofactor.111 Taken together, these results suggest that activation of factor VIII resulting in limited proteolytic cleavage is accompanied by changes in the protein conformation.
Finally, the cellular surface profoundly affects the procoagulant activity of factor VIII. Negatively charged phospholipids are usually confined to the inner leaflet of cellular membranes and are exposed upon cellular lysis at sites of injury.
Formation of the Xase Complex
Interaction with Factor IXa
The proteolytically activated form of factor VIII, factor VIIIa, serves as a cofactor for the serine protease factor IXa in the conversion of factor X to factor Xa. This complex of enzyme and cofactor, assembled on an anionic phospholipid surface, is referred to as the intrinsic factor Xase. The phospholipid surface is primarily involved in reducing molecular interactions to a 2-D space, thereby markedly decreasing the Km for factor X. The role of surface also appears to contribute to the Kcat effect.116 The association of factor VIIIa and factor IXa is complex and not fully understood. This interaction has been studied by homology modeling, analysis of naturally occurring hemophilia A and B mutations, mutagenesis, cross-linking, and inhibition with synthetic peptides. All these studies came to the conclusion that the interaction sites are localized in the A2 domain (residues 558 to 569, residues 484 to 509, residue Asp527, and residues 708 to 715) and the A3 domain (residues 1811 to 1818).114,117,118,119,120,121,122,123,124 The molecular model of factor VIII46, although not the activated form, mapped the potential factor IXa binding sites on the A2 domain at residues 558 to 665 and the region around Asp712 and in the A3 domain in the region of residues 1811 to 1818. All these regions appear on the model to be solvent exposed and thus likely accessible to binding factor IXa (see further FIGURE 12.7).
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


