Eye and Orbit
Tumors of the eye and orbit are rare. The American Cancer Society estimates that in 2011 there will be 2,570 new cases with 240 deaths.1 Male to female incidence is similar, with 1,270 cases occurring in men and 1,300 in women. In adults, melanoma is the most common primary intraocular cancer, followed by lymphoma, while in children retinoblastoma is the most common tumor, followed by medulloepithelioma. Metastases, or secondary intraocular tumors, are more common than primary tumors and typically come from breast or lung cancers.2 Numerous other tumors such as rhabdomyosarcoma, optic nerve glioma, conjunctival tumors, and eyelid carcinomas also occur in the orbit. Radiation therapy has been effective in the treatment of many of these tumors and can be delivered externally or by brachytherapy, depending on the clinical situation, and can be used exclusively or in concert with other treatments such as surgery or chemotherapy. Technological advances such as intensity modulated radiation therapy (IMRT), proton beam therapy, and stereotactic radiotherapy have a key role in management of these tumors given the anatomy proximity of structures in this location. This chapter will outline the most relevant malignant and benign conditions of the eye and orbit with emphasis on radiotherapeutic management.
 ANATOMY
ANATOMY
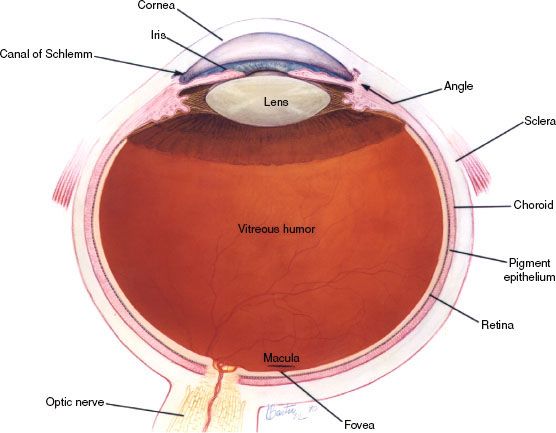
The eye is not an exact sphere, but rather a fused two-piece unit. The smaller, more curved frontal unit is the cornea and is linked to the larger unit called the sclera. The corneal segment is typically about 8 mm in radius. The sclera constitutes the remainder of the eyeball, with a radius of approximately 12 mm. The cornea and sclera are connected by the limbus. The iris and the pupil are seen instead of the cornea due to the cornea’s transparency. The area opposite the pupil, the fundus, shows the characteristic pale optic disc or papilla, where vessels enter the eye and optic nerve fibers depart the globe. Dimensions of the globe differ among adults by only 1 or 2 mm. The vertical measure, generally less than the horizontal distance, is about 24 mm in adults. The eye is made up of three coats or tunics, enclosing three transparent structures (Fig. 38.1). The outermost layer is composed of the cornea and sclera. The middle layer consists of the choroid, ciliary body, and iris. The innermost is the retina, where the blood supply is from the vessels of the choroid as well as the retinal vessels. The lens is suspended to the ciliary body by the suspensory ligament, made up of fine transparent fibers.
 RADIATION TOLERANCE OF OCULAR STRUCTURES
RADIATION TOLERANCE OF OCULAR STRUCTURES
Eyelid
Acute radiosensitivity of eyelid skin is comparable to skin at other sites, and loss of eyelashes may occur at doses as low as 20 Gy using standard fractionation.3 Late effects can include telangiectasia and atrophy. Xerophthalmia can result from doses as low as 24 to 26 Gy and may be the result of dysfunction of the Meibomian glands, lacrimal acinar cells, or both.4 Significant xerophthalmia can cause corneal desiccation and pain. It is generally believed that smaller fraction sizes, longer treatment schedules, and smaller volumes will reduce late effects, but this must be weighed against the potential for ocular trauma when using shielding on a daily basis.5 Optimum management of eyelid toxicity includes cleanliness, dressings for moist desquamation, healing time, and artificial tears for Meibomian gland dysfunction.
Conjunctiva
Acute conjunctivitis is common with doses ≥30 Gy and secondary bacterial or rarely viral infections may occur.6 Conjunctivitis can be reduced by treating with an open eye with megavoltage equipment if the clinical situation permits. Treatment of acute conjunctivitis involves artificial tears to relieve symptoms and treatment of an underlying infection when indicated.
Lacrimal System
The lacrimal gland system includes the main lacrimal glands, accessory lacrimal glands, and lacrimal duct system, and symptoms of dryness can occur if any of these structures receive radiation. The most concerning late effect is dry-eye syndrome, where patients can experience tearing, redness, discharge, foreign body sensation, blurred vision, and photophobia. Moderate-dose orbital radiation therapy (RT) (30–45 Gy) can cause dry-eye syndrome 4 to 11 years after treatment, while higher doses (>57 Gy) can produce it in 9 to 10 months.7 Lacrimal shielding should be used if tumor control will not be compromised, and shielding the accessory lacrimal glands may reduce toxicity if the main gland is irradiated. IMRT can help minimize the risks of RT-induced xerophthalmia, and prophylactic nasolacrimal duct intubation with silicon tubing may be considered in high-risk patients.8,9 Treatments available for RT-induced xerophthalmia include topical lubricants, moist chamber goggles, punctual occlusion with plugs, or tarsorrhaphy.
FIGURE 38.1. Normal eye anatomy. (Courtesy of National Eye Institute, National Institutes of Health.)
Cornea
Although RT can directly injure the cornea, most acute corneal toxicity results from loss of the tear film with secondary keratitis sicca. Punctate epithelial erosions are common after conventionally fractionated RT doses of 30 to 50 Gy.10 They typically subside within several weeks but can persist for years. At higher doses, corneal edema (40–50 Gy) or perforation (60 Gy) may occur, causing pain, tearing, foreign body sensation, or reduced vision. Corneal toxicity can be reduced by using megavoltage equipment and reducing the surface dose or by using careful RT planning to minimize corneal irradiation so long as the tumor dose is not compromised. Commercially available eye shields may also be used, particularly for electron beam RT.11 There are few published data on management of corneal toxicity from RT. Close ophthalmologic follow-up is recommended so topical treatment with steroids or antibiotics can be administered when indicated.
Iris
The iris is relatively radioresistant and thus, acute iritis is rare. However, persistent iritis, with symptoms such as pain, red eye, and blurred vision, has been observed after hypofractionated RT doses of 30 to 40 Gy and after doses ≥70 Gy given with conventional fractionation.12 A problematic late effect of the iris is neovascular glaucoma, where patients may present with ocular pain, headache, photophobia, decreased vision, and redness.13 Risk factors include higher radiation dose, diabetes, vitreous hemorrhage, and retinal detachment. Where possible, techniques that spare the anterior chamber should be used. The primary treatments for iritis and neovascular glaucoma are topical steroids and cycloplegic drops, respectively. Laser panretinal photocoagulation or peripheral cryotherapy may prevent or slow the progression of glaucoma if performed early. However, in many cases more aggressive intervention such as trabeculectomy may be required. Occasionally progression to a blind, painful eye may necessitate enucleation. Novel therapies such as intravitreal bevacizumab as an adjunctive treatment to retinal ablative procedure appear to be promising for the management of iris neovascularization associated with neovascular glaucoma.14
Lens
Age at time of treatment, total dose, and fractionation contribute to cataract formation.15,16 Hall et al.15 estimated an increased risk of approximately 50% for 1-Gy exposure to the lens during childhood. In adults, higher doses are associated with cataract: after 2.5 to 6.5 Gy, the latent period is 8 years with a 33% of progressive cataract, while after 6.51 to 11.5 Gy, the latent period is 4 years, with a 66% risk.16 Cataract risk can be reduced by using customized lens shields and lens-sparing techniques or by using fully fractionated RT schedules. IMRT may be used to reduce cataract risk by reducing overall lens dose and relative fraction size. The definitive treatment for RT-induced cataract is surgery, which yields excellent results.
Retina
Radiation retinopathy is a late effect of RT that typically presents 6 months to 3 years after treatment, although cases have been reported as long as 15 years after therapy.17 Patients may be asymptomatic or may complain of floaters or reduced visual acuity, and clinical signs include microaneurysms, telangiectasia, hard exudates, cotton wool spots, and neovascularization. The threshold dose for retinal damage is usually considered to be 30 to 35 Gy. In a Cochrane database review of RT for macular degeneration, no retinopathy or optic nerve damage was reported in 1,154 patients treated with doses up to 24 Gy.18 The risk of retinopathy increases dramatically when doses exceed 50 Gy using standard fractionation.19 Incidence as well as severity are also increased by coexistent diabetic retinopathy, hypertension, collagen vascular disease, simultaneous chemotherapy, and pregnancy.20 No proven therapy exists for radiation retinopathy, although local treatments such as photocoagulation may improve symptoms and there is interest in intravitreal bevacizumab.21
Optic Nerve
Like the retina, the optic nerve manifests toxicity months or years after RT, with a peak incidence at 18 months.22 Radiation-induced optic neuropathy (RION) has a variable presentation and is related to the nerve fibers most affected, usually causing visual field defects. In a recent review by the Mayo Clinic, risk of RION was almost zero with conventionally fractionated doses ≤50 Gy and the occurrence of RION is still rare with a maximum dose <55 Gy. The risk of RION increases from 3% to 7% at 55 to 60 Gy and is substantial (>7% to 20%) at doses >60 Gy.23 Parsons et al.,24 in a series of 131 patients (215 optic nerves) treated with RT for extracranial head and neck tumors, found no RION in nerves that received <59 Gy. Fraction size was of primary importance; in cases where >60 Gy was received, fraction size was more important than total dose in producing RION. The 15-year actuarial risk was 11% when fraction size was <1.9 Gy compared with 47% when fraction size was >1.9 Gy. A significant exception in development of RION appears to exist for patients treated for pituitary tumors, where toxicity has been reported at doses as low as 46 Gy in 1.8-Gy fractions.23 Because there is no known effective therapy for RION, efforts must be made to minimize optic nerve dose through sophisticated treatment planning and delivery with an effort to minimize treatment volume, total dose, and especially dose per fraction received.
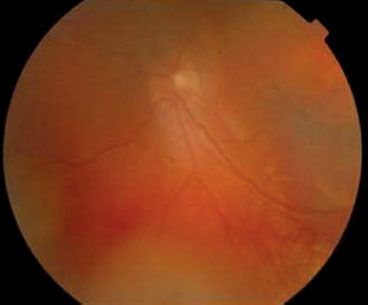
FIGURE 38.2. Diffuse choroidal hemangioma with exudative retinal detachment. (From Kubicka-Trza˛ska A, Kobylarz J, Romanowska-Dixon B. Ruthenium-106 plaque therapy for diffuse choroidal hemangioma in Sturge-Weber syndrome. Case Rep Ophthalmol Med 2011, art. 785686.)
 MANAGEMENT OF BENIGN OCULAR DISEASES
MANAGEMENT OF BENIGN OCULAR DISEASES
Pterygium
Pterygium is a benign growth of fibrovascular tissue on the conjunctiva that can cause irritation, erythema of the cornea, and obstructed vision in advanced cases. The exact cause of pterygium is unknown, but it is associated with excessive exposure to wind, sunlight, or sand. It has also been postulated that ultraviolet light exposure may increase the risk of pterygium development.25 At present, no reliable medical treatment exists to reduce or even prevent pterygium progression. If symptoms are severe, the only definitive treatment is surgical removal, which requires removal of the head, neck, and body of the pterygium. Without adjuvant treatment, surgical resection alone, commonly referred to as bare sclera excision, carries recurrence rates of 20% to 80%.25–27 Therefore, adjunctive measures are recommended, which can be broadly classified as medical methods, beta-irradiation, and surgical methods. Intraoperative and postoperative mitomycin-C are the most commonly used medical adjuncts, and recurrence rates of 3% to 37% have been reported.28 However, topical chemotherapy is not commonly used out of concern for late complications, including scleral sclerosis, infectious scleritis, perforation, or endophthalmitis, all of which can impair vision.29 Beta-irradiation has a historical role in management of pterygium. In a prospective randomized study, 96 eyes with primary pterygium received beta-irradiation with a strontium-90 after resection or sham radiation27 Local control was 93.2% for the irradiated group versus 33.3% for the sham radiation group. Like topical chemotherapy, however, beta-irradiation has generally been abandoned due to the risk of sight-threatening complications such as scleral necrosis, infectious scleritis, corneal perforation, and endophthalmitis.30 Surgical adjuvant therapy is thus the mainstay of management for primary and recurrent pterygium. Conjunctival autografting is generally regarded as the procedure of choice because of its efficacy and long-term safety, with recurrence rates of 2% to 39% without the attendant sight-threatening complications of topical chemotherapy or beta-irradiation.31 Other surgical techniques include amniotic membrane transplantation, limbal conjunctival transplantation, and cultivated conjunctival translation, but none appear to be more effective than conjunctival autografting.32–34
Choroidal Hemangiomas
Choroidal hemangiomas are benign vascular tumors of the choroid. Although probably congenital in all cases, they are frequently undetected until after the second decade of life and can have a wide range of clinical features and treatment options.35 These tumors are characterized as either circumscribed or diffuse type. Most circumscribed choroidal hemangiomas are first noted when they produce visual symptoms caused by accumulation of serous subretinal fluid, degenerative changes in the macular retina, or both. In contrast to circumscribed choroidal hemangiomas, the diffuse variety is large and is associated with manifestations of the Sturge-Weber syndrome.35 Diffuse tumors are usually diagnosed in young patients either due to examination of the fundus prompted by a facial hemangioma or due to visual impairment secondary to serous retinal detachment or hyperopic amblyopia (Fig. 38.2). The clinical course of either form of choroidal hemangioma is highly variable, with visual impairment ranging from none to total blindness. Neither variety of choroidal hemangioma metastasizes or transforms to malignancy. Therefore, the primary indication for treatment is loss of visual acuity.
Treatment alternatives include laser photocoagulation, thermotherapy, photodynamic therapy, and radiotherapy. Photocoagulation is beneficial for circumscribed lesions, but diffuse lesions have high recurrence rate and retinal damage is possible.34 Low to moderate dose radiation using lens-sparing external beam photon irradiation, episcleral plaque therapy, proton beam therapy, and stereotactic radiotherapy have been used in the treatment of choroidal hemangioma.35,36 Total doses of 18 to 30 Gy delivered in 10 to 18 fractions of external beam photon radiation therapy can result in partial flattening of the hemangioma, resorption of subretinal fluid, and reattachment of the retina within 6 to 12 months.36,37 Heimann et al.37 reported no recurrence of subretinal fluid with at a mean follow-up of 3.6 years. In another series, Kivela et al.38 found subretinal fluid had reaccumulated in only 1 of 12 patients treated with follow-up of 66 months, thus illustrating response durability. In very advanced cases with retinal detachment, a higher dose of 36 Gy in fractions of 1.8 Gy has been described and appears to be efficacious, but clearly larger series with longer follow-up are needed to describe response durability and delayed side effects, including retinopathy and cataract formation.37,38 Lens-sparing techniques, including three-dimensional conformal radiation therapy with computed tomography (CT) planning, should be considered to reduce the incidence of cataract formation39,40 Brachytherapy has also been used for choroidal hemangioma treatment and particularly for circumscribed lesions, given the focused dose distribution of brachytherapy. This treatment usually achieves resolution of subretinal fluid and reattachment of the retina with preservation of pretreatment visual acuity.41 Radiation dose varies with the isotope, but in one study using iodine-125, a target dose of 48 Gy to the apex was prescribed and tumor regression was noted in eight of eight cases.41 More recently results of proton beam therapy have been reported from investigators at the Institut Curie. In a series of 71 cases with circumscribed choroidal hemangioma treated with 20 cobalt gray equivalent (CGE), retinal reattachment occurred in all cases and a completely flat scar was obtained in 91.5%.42 The main complications during the surveillance period were cataract (28%) and radiation-induced maculopathy (8%). No cases of eyelid complications or neurovascular glaucoma were observed.
Capillary Hemangioma
Capillary hemangiomas are benign endothelial cell neoplasms that rarely occur on the eyelids or skin of the orbit. Retinal capillary hemangiomas may represent a component of the von Hippel-Lindau syndrome, and lesions of the face that occupy the distribution of the trigeminal nerve can be a component of Sturge-Weber syndrome. The natural history of these lesions is usually spontaneous regression over 3 to 4 years, therefore, conservative management is the treatment of choice.43 Occasionally, however, lesions may be large enough to obstruct vision and amblyopia may occur. Treatment options then include corticosteroids, interferon alfa-2a, laser therapy, embolization, immunomodulators, surgery, and systemic propranolol.44 Radiation therapy in the management of capillary hemangiomas is primarily of historical interest, with historical studies showing that doses in the 16 to 20 Gy range provide effective local control.45
Orbital Pseudotumor
Orbital pseudotumor is a rare inflammatory process that affects the soft tissue components of the orbit. Clinically, it presents in the fourth and fifth decades with signs and symptoms including proptosis, swelling, increased orbital pressure, and restricted ocular motion.46 The diagnosis is based on history, imaging of the orbit, and pathologic examination of tissue in accessible lesions. Characteristic imaging features include extraocular muscle enlargement, optic nerve thickening, and inflammation of retrobulbar adipose tissue. Treatment options for orbital pseudotumor include corticosteroids, external beam radiation therapy (EBRT), immunotherapy, chemotherapy, and surgery; corticosteroids are typically used as primary treatment.47 EBRT is usually reserved for cases that are refractory to corticosteroid treatment, with local control rates of 67% to 83%.48,49 Matthiesen et al.50 recently described 20 orbits treated with EBRT to a median dose of 20 Gy with follow-up of 16.5 months and found 87.5% had improved symptoms or reduction of corticosteroid dose and 56% were able to discontinue steroid treatment completely. Based on these observations, EBRT appears to achieve durable control in orbital pseudotumor and is an effective strategy in patients who respond poorly to medical treatment.
Thyroid-Associated Orbitopathy
Thyroid-associated orbitopathy (TAO), frequently termed Graves’ ophthalmopathy, is part of an autoimmune process that can affect the orbital and periorbital tissue, the thyroid gland, and, rarely, the pretibial skin or digits (thyroid acropathy).51–52,53 Although the use of the term thyroid ophthalmopathy is pervasive, the disease process is actually an orbitopathy in which the orbital and periocular soft tissues are primarily affected with secondary effects on the eye. TAO may compromise a patient’s vision by causing diplopia, decreased ocular motion, exposure keratitis, or optic neuropathy. A variety of treatments exist including thyroid hormone regulation, corticosteroids, external beam radiotherapy, or surgical decompression.54 Radiation therapy historically has been used in cases refractory to medical therapy. Doses of 20 Gy using standard fractionation have been effective in ameliorating symptoms and providing durable control, with one series reporting 87% of patients having improvement in symptoms.55 However, prospective studies examining the use to radiation therapy for TAO suggest that its role is unclear. Nine randomized controlled trials have tested orbital RT in 465 patients with TAO, although studies differed with respect to severity of TAO on trial entry, radiation dose and fractionation, and inconsistency in the use of concurrent corticosteroids.56–64 Three of these studies, which compared orbital RT with sham control, reported only minor improvement in outcome. Mourits et al.60 randomized 59 patients to either 20 Gy in 10 fractions to both orbits or sham RT and found improved globe motility and elevation, but no difference in change in lid fissure, soft tissue swelling, proptosis, or clinical activity score (a measure of disease activity and propensity to progress). The authors concluded that RT should be used only for motility impairment. Gorman et al.64 randomized 42 subjects to orbit RT to one orbit and sham RT to the other followed by the reverse therapy 6 months later. No clinically or statistically significant differences were observed at 6 months. At 12 months, muscle volume and proptosis were slightly more improved in the orbit that was first treated. Prummel et al.63 conducted a trial where 88 patients with mild TAO were randomly assigned to bilateral RT or sham RT. Irradiation was effective in improving motility and decreasing severe diplopia, but no differences in health-related quality of life were detected, although lack of complete quality-of-life data on 58% of subjects resulted in low power to detect a difference. Complications of orbital RT are rare but not insignificant. Even in the absence of known diabetes mellitus, the risk of definite retinopathy is 1% to 2% in the first 10 years after treatment.65,66 Based on these randomized data, the role of RT for TAO is thus controversial. In a 2008 report on orbital RT for TAO by the American Academy of Ophthalmology, the investigators concluded that while extraocular motility may improve with RT, the evidence of treatment effect is mixed in clinical trials, and future studies are needed to determine if improved motility translates into improved quality of life.67 Radiation technique for TAO usually involves treatment of both orbits with parallel opposed lateral portals with low energy (6 MV) photons. A downward 5-degree tilt or use of half-beam block should be used to avoid direct radiation of the contralateral lens.
 OCULAR AND ORBITAL MALIGNANT TUMORS
OCULAR AND ORBITAL MALIGNANT TUMORS
Metastatic Carcinoma to the Uvea
Tumor metastases to the eye are more common than primary ocular cancers, with the uvea representing the most common site affected.68 Shields et al.2 described a comprehensive series of usual metastases and found that within the uvea, 88% of metastases are to the choroid (Fig. 38.3), followed by metastases to the iris (9%) and ciliary body (2%). This large difference is thought to be due to the distribution of blood supply, which heavily favors the choroid as compared to the iris or ciliary body. The most common primary cancer sites for uveal metastasis in males were lung (40%), gastrointestinal (9%), and kidney (8%). The primary site was unknown at the time of presentation in 29% of males. In females, the most common sites included breast (68%), lung (12%), and other (4%). Metastases can be either unilateral or bilateral. In a study of 264 patients with uveal metastases from breast cancer reported by Demirci et al.,69 62% of patients had unilateral metastasis at presentation. Patients with breast cancer metastatic to the uvea show survival rates of 65% at 1 year, 35% at 3 years, and 24% at 5 years.
Numerous treatment options exist for choroidal metastases, including systemic chemotherapy, hormonal therapy, EBRT, brachytherapy, or photodynamic therapy. Individualized treatment should be considered, taking into account disease extent and life-expectancy with the primary goal of maintaining visual acuity. As systemic treatments for metastatic cancers improve, clinicians may be faced with treatment of symptomatic choroidal metastases. For cases with multifocal or diffuse presentations, EBRT has been effective with doses in the range of 20 to 40 Gy. Rosset et al.70 described 58 patients (88 eyes) treated with external radiation with a median dose of 35.5 Gy (range 20–53 Gy) in 10 to 30 fractions. Various techniques were used and lens sparing was used when possible. Visual acuity improved in 62% of patients, with significantly better results when doses >35.5 Gy were used. Five complications were noted, including three cataracts, retinopathy in a single patient who underwent biopsy, and one case of glaucoma from subretinal hemorrhage. In cases of unilateral disease, the authors noted no cases of new contralateral lesions when bilateral radiation was performed, but did describe new contralateral lesions in 3 of 26 patients when unilateral technique was used. They have thus recommended bilateral irradiation even in cases of unilateral disease (Fig. 38.4).
Plaque brachytherapy is usually reserved for solitary metastases. This modality offers precise, controlled radiation delivery to the eye and requires only 3 to 4 days of treatment, which is an important consideration in uveal metastases because many patients have limited survival expectancy. Key clinical factors to consider for plaque therapy include the size and thickness of the lesion, the distance of the lesion from the optic nerve, and the distance of the lesion from the foveola. Investigators from the Wills Eye Hospital studied 36 patients who received plaque radiotherapy either as primary or salvage treatment (after external radiation) for uveal metastases.71 The mean duration of treatment was 86 hours and mean dose to the apex and base of the tumor was 68.8 Gy and 235.6 Gy, respectively. Tumor regression was documented in 94% of cases with mean follow-up of 11 months. In six cases where plaque therapy was used after suboptimal response to external irradiation, five eyes were successfully salvaged. Complications from plaque radiotherapy are similar to those of EBRT, including dryness, radiation retinopathy, papillopathy, and cataract, but these side effects are uncommon, especially given the short life-expectancy of many patients. The investigators concluded that plaque radiation is an effective, time-efficient method for treatment of selected solitary uveal metastases. Thus, both EBRT and plaque therapy are effective therapies for uveal metastases, with the optimal treatment depending on the extent of intraocular disease, symptoms, and overall condition and prognosis of the patient. Close collaboration of ophthalmologists, radiation oncologists, and medical oncologists is imperative to develop an appropriate treatment strategy.
FIGURE 38.3. Fundus fluorescein angiography of choroidal metastasis from non–small cell lung cancer showing hyperfluorescence from the surface of the choroidal tumor in its late phase with the accumulation of subretinal fluid. (From Singh A, Singh P, Sahni K, et al. Non-small cell lung cancer presenting with choroidal metastasis as first sign and showing good response to chemotherapy alone: a case report. J Med Case Rep 2010;4:185.)
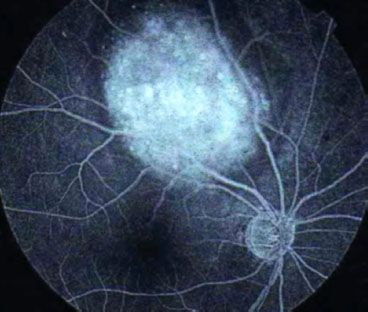
FIGURE 38.4. Bilateral radiation therapy for uveal metastases in a patient with widely metastatic lung cancer.
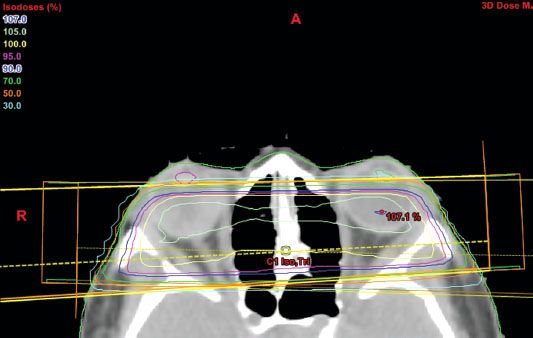
FIGURE 38.5. A: Iris melanoma located at the inferonasal aspect of the eye. B: Typical choroidal melanoma with associated nonrhegmatogenous retinal detachment. (From Papastefanou VP, Cohen VML. Uveal melanoma. J Skin Cancer 2011, art. 573974.)
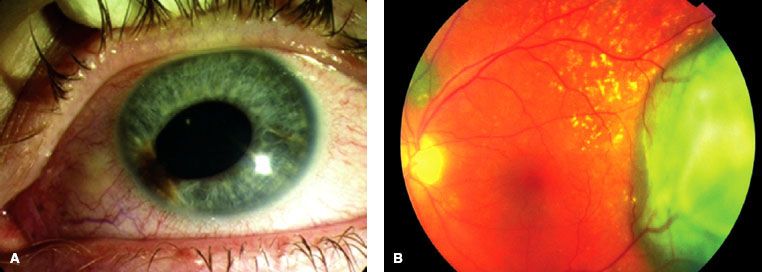
Malignant Melanoma of the Uvea
Uveal melanomas represent <5% of all malignant melanomas with approximately 1,400 new cases per year in the United States.72 These tumors may arise from any of the three parts of the uvea and are sometimes referred to by their location, such as iris melanoma (Fig. 38.5A), ciliary body melanoma, or choroidal melanoma (Fig. 38.5B). True iris melanomas, originating from within the iris as opposed to originating elsewhere and invading the iris, are distinct in their etiology and prognosis, such that the other tumors are often referred to collectively as posterior uveal melanomas. Although uveal melanoma is rare among nonwhites, the role of sunlight and other environmental factors is unknown.73 Detection of uveal melanoma is often by routine examination with or without symptoms; a study from the United Kingdom showed that 45% of patients were asymptomatic when their tumor was detected.74 Advances in diagnostic techniques such as binocular indirect ophthalmoscopy, angiography, and B-scan ultrasonography have aided ocular oncologists in the evaluation and management of patients with uveal melanoma.75,76 Staging of iris differs from ciliary body and choroidal lesion, with thickness being of primary importance in choroidal and ciliary body tumors (Table 38.1).
Several treatment options exist for uveal melanoma. Most patients are treated with the goal of eradication of disease and long-term survival. There is evidence that local tumor recurrence is associated with increased mortality.77 Factors predictive of local recurrence include epithelioid cell type, large tumor size, and posterior tumor extension.78 When possible, curative treatment should be sought with the goal of preserving vision with acceptable cosmesis. In patients with known metastatic disease, the main objective is to preserve vision and remove any threat of the eye becoming painful within the patient’s life-expectancy. Many factors influence treatment selection, including tumor size, location, and extent; secondary effects, such as cataract; concurrent ocular disease, such as diabetic retinopathy; the patient’s general health and life-expectancy; and the cost and duration of the treatment.79 In cases where maintaining useful vision is feasible, organ-preservation therapy should be considered, and thus radiation therapy, delivered either externally or by brachytherapy, has been instrumental in the primary management of uveal melanoma.
Observation
It is not uncommon for indeterminate pigmented uveal tumors to be observed without treatment until growth is documented. The probability of malignancy can be estimated according to tumor thickness, serous retinal detachment, orange pigment, and symptoms.80 Patients and clinicians can then make a combined decision on when to commence therapy after discussion of risks and benefits.
Enucleation
Enucleation was traditionally the standard of care for choroidal melanoma since the late 19th century, but its effectiveness in improving survival has never been clearly demonstrated.81 A study from the Helsinki University Hospital looked at the long-term prognosis of patients treated by enucleation between 1962 and 1981 and found death was attributable to melanoma in 61% of cases.82 They also reported that cause-specific mortality (CSM) increased with longer follow-up. The 5-, 15-, 25-, and 35-year CSMs were 31%, 45%, 49%, and 52%, respectively, thus illustrating that a substantial number of patients die of metastatic disease more than 5 years after enucleation. The desire to improve survival and preserve vision stimulated the development of alternative, organ-preserving therapies for uveal melanoma. Still, enucleation is required in a subset of patients, either because tumor is too extensive at presentation or because complications of conservative therapy, namely vision loss, would be too high. General guidelines for enucleation include tumor diameter >17 mm, thickness >6 to 7 mm, involvement of the optic disc, invasion of more than 30% of the iris, ciliary body, or angle, retinal perforation, or poor general health of the patient.79 It was once postulated that surgical manipulation during enucleation could disseminate tumor cells into blood vessels and thereby increase the possibility of metastatic spread.81 This spawned an interest in preoperative radiotherapy in an effort to prevent dissemination, but a more recent study showed no improvement in survival when preenucleation radiation was used.83
Endoresection
Transretinal endoresection is controversial, mainly because of fears of seeding tumor cells, but has been advocated for juxtopapillary tumors up to 10 mm in diameter.79 The operation involves vitrectomy; tumor removal with a vitrector either via a retinotomy or after lifting a retinal flap; fluid–air exchange to drain any subretinal fluid; endolaser photocoagulation to destroy any residual tumor in the sclera and to achieve retinopexy; air–silicone exchange; and, if possible, adjunctive brachytherapy.84 Given the seemingly heightened risk of tumor seeding with endoresection, some investigators have recommended preoperative stereotactic radiation, but this practice is similarly controversial.85 Endoresection carries approximately 10% risk of local recurrence, which can arise from microscopic disease in the scleral bed or at the margins of resection.84
Transscleral Resection
Transscleral local resection has been promoted for tumors >6-mm thick in patients highly motivated to retain vision and in patients with severe exudative retinal detachment after radiotherapy.79 The procedure carries substantial operative risk. Choroidectomy and cyclochoroidectomy, for example, require hypotensive anesthesia with systolic blood pressure lowered to approximately 40 mm Hg.86 The operation involves the preparation of a lamellar scleral flap, ocular decompression by limited pars plana vitrectomy, resection of the tumor together with the deep scleral lamella, suturing of the scleral flap, intraocular injection of balanced salt solution, and adjunctive brachytherapy either at the end of the operation or a several weeks later. This procedure has approximately 30% incidence of local relapse when performed exclusively, but this probability declines to approximately 10% when perioperative brachytherapy is delivered.87
TABLE 38.1 UVEAL MELANOMA STAGING


Transpupillary Thermotherapy
Photocoagulation of choroidal melanoma using brief flashes of light has also been investigated, but was superseded by low-energy, long-duration krypton laser photocoagulation, which has greater penetration.88 This modality was surpassed by transpupillary thermotherapy, in which 1-minute applications of 3-mm spots of low-energy diode laser are administered to the tumor and the surrounding choroid.89 The objective is not immediate thermoablation, but rather heating the tumor by only a few degrees so that following treatment, tumor regression occurs slowly, often resulting in a white scar. This technique may be used for indeterminate choroidal tumors. Some advocates of transpupillary thermotherapy recommend adjunctive brachytherapy, while others have attempted phototherapy without radiotherapy.90
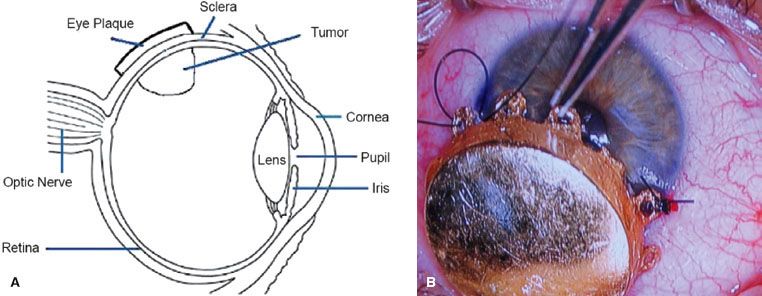
Plaque Brachytherapy
Plaque brachytherapy is the mainstay of treatment in many centers, with iodine-125 and ruthenium-106 being the most common isotopes, although palladium-103 has also been used effectively.91 Iodine emits γ-rays, which have a range sufficient for tumors up to 8- to 10-mm thick, while ruthenium delivers beta-particles that have a more limited range, which is suitable for tumors up to approximately 5 mm. The general objective with all plaques is to deliver approximately 80 Gy to the tumor apex by fixing the plaque in the exact location of the tumor (Fig. 38.6). Computer modeling has been developed, as seen in general radiotherapy planning, to create a three-dimensional model of the eye and determine the appropriate treatment time and estimated dose to the optic nerve, macula, and lens.92 The largest prospective experience of patients treated by plaque brachytherapy was conducted by the Collaborative Ocular Melanoma Study (COMS) group. From 1986 to 2003, COMS conducted two multicenter trials of brachytherapy with iodine-125 versus enucleation in selected patients with choroidal melanoma. Long-term results were subsequently published in COMS report 28 in 2006, which evaluated 1,317 patients.93 Eligible patients had unilateral choroidal melanoma with an apical height of 2.5 to 10 mm and a maximum basal tumor diameter (MBTD) of 16 mm. Patients whose tumors were contiguous with the optic disc were ineligible, as were patients with metastases from melanoma or another malignancy. Patients were followed for 5 to 15 years, and within 12 years after enrollment, 471 of 1,317 (36%) had died. Overall cause-specific mortality at 5 and 10 years for both treatment arms were 19% and 35%, respectively. Cumulative all-cause mortality was 43% in the iodine-125 arm and 41% in the enucleation arm, indicating that with long-term follow-up no survival difference existed between plaque brachytherapy and enucleation. Age older than 60 years and MBTD >11 mm were the primary predictors of time to death from all causes and death with melanoma metastases. Local control was excellent in the COMS series, with only 12.5% of patients requiring salvage enucleation.94 Visual acuity after plaque brachytherapy depends on a number of features. Shields et al.95 examined 1,106 patients with visual acuity of 20/100 or better who underwent brachytherapy for uveal melanoma. They found 34% of patients had poor visual acuity at 5 years and 68% at 10 years, defined as 20/200 to no light perception. Factors adversely affecting visual acuity were age over 60 years, poor vision at baseline, increasing tumor thickness (>8 mm), proximity to the foveola of <5 mm, recurrent tumor, subretinal fluid, and history of diabetes or hypertension. Best results were obtained in eyes with small tumors outside a radius of 5 mm from the optic disc and foveola. Whether improvements in tumor imaging and localization and radiation planning will improve functional results will be determined with further investigation.
FIGURE 38.6. A: Eye anatomy and plaque placement in location of tumor. (From Chaudhari S, Deshpande S, Anand V, et al. Dosimetry and treatment planning of Occu-Prosta I-125 seeds for intraocular lesions. J Med Phys 2008;33:14–18.) B: Plaque brachytherapy for melanoma of the iris. (From Khan MK, Khan N, Almasan A, et al. Future of radiation therapy for malignant melanoma in an era of newer, more effective biological agents. Onco Targets Ther 2011;4:137–148.)
Proton Beam Therapy
Proton beam therapy may be used to treat uveal melanoma and is usually indicated for tumors that extend close to or are contiguous with the optic disc, referred to as parapapillary and peripapillary tumors, respectively.96 Large tumors and lesions of the iris and ciliary body may also be candidates for this modality as are patients who are not fit for operative therapy.79 Because a proton beam delivers a homogeneous dose to tumor and has a sharp edge, a high tumor dose can be delivered with relative sparing of the optic nerve. The decision to use proton therapy over other forms of external therapy, such as helium ions or stereotactic radiotherapy, often depends on the availability of treatment facilities in addition to clinical factors. One of the largest experiences in proton beam therapy for uveal melanoma is from the Harvard Medical School.97 Treatment planning involves intraoperative examination by transillumination or indirect ophthalmoscopy, and the edges of the tumor are delineated by four tantalum rings sutured to the sclera. For tumors of the ciliary body and peripheral choroid, surgery is not performed; transillumination is instead used to define tumor margins in relation to the iris and cornea. Radiation planning is done with an interactive three-dimensional computer system to define the beam aperture and range modulation needed to adequately encompass the tumor and a 1.5-mm margin is included to allow for motion during treatment, setup error, and possible microscopic extension.98 Patients receive a total dose of 70 CGE, which is delivered in 5 equal fractions over 7 to 10 days (63.6 proton Gy é 1.1 relative biological effectiveness = 70 CGE).97 The Harvard group recently reported long-term follow-up in a series of 573 patients with peripapillary and parapapillary melanomas with median surveillance of 96.3 months.96 Local recurrence was rare, with 5- and 10-year local recurrence of 3.3% and 6%, respectively, and similarly high rates of local control have been observed in a larger series from the same institution.99 Enucleation rates were 13.3% and 17.1% at 5 and 10 years after treatment, respectively. Of 450 patients with baseline visual acuity of 20/200 or better, two-thirds had visual acuity <20/200 years after treatment, although 56% could count fingers. The most common complications were radiation maculopathy and papillopathy. By 3 years the cumulative rate of both complications was 49%, and by 10 years, this increased to 61% for papillopathy and 68% for maculopathy. The visual outcome after proton therapy depends on the height of the tumor and its location relative to the fovea and optic nerve.99 Anterior segment complications, such as rubeosis iridis and neovascular glaucoma, are the most serious, but occur less frequently, with each occurring in approximately 15% of cases at 5 years postradiation.100 These results indicate that although visual acuity is compromised with proton beam therapy, particularly in patients with juxtapapillary lesions, some preservation is possible and eye conservation is likely with extremely low rates of local recurrence.
Stereotactic Radiotherapy
Stereotactic radiation can be delivered by linear accelerator (LINAC) or by specialized devices for focused radiation such as the Leksell Gamma Knife (Elekta, Norcross, GA), which provides focused radiation with a multitude of sources. Because gamma knife treatment is usually done in a single fraction, the term radiosurgery is applied, whereas for LINAC-based treatment, single fraction or multifraction treatment is possible. Gamma knife radiosurgery (GKR) for uveal melanoma was first introduced in 1998 by investigators at the Indiana University School of Medicine.101 Nineteen patients with uveal melanoma were treated to a dose of 40 Gy prescribed to the 50% isodose line. With median follow-up was 40 months, 3- and 5-year overall survival rates were 86 and 55%, respectively. The 3- and 5-year tumor control rates were both 94%. Six of the 19 treated patients (32%) developed distant metastasis 31 to 75 months after GKR. Of the 19 patients treated, 2 had improved, 4 had stable, and 13 had worse vision in the treated eye. Similar results were reported in a larger series of 78 patients from investigators in Milan.102 The dose was adjusted over the treatment period: 7 patients received 50 Gy at the 50% isodose line (1994–1995), 21 patients received 40 Gy to the 50% isodose line (1995–1999), and 47 patients received 35 Gy to the 50% isodose line (2000–2006). Local tumor control was achieved in 91.0% of patients and the eye retention rate was 89.7%. A significant relative reduction of visual acuity was observed during follow-up. The most frequently encountered complications were exudative retinopathy (33.3%), neovascular glaucoma (18.7%), radiogenic retinopathy (13.5%) and vitreous hemorrhage (10.4%).
One theoretical disadvantage of single-dose stereotactic radiosurgery is the potential for complications, which has prompted investigators to explore fractionated stereotactic radiotherapy (SRT) treatment of uveal melanoma.103 In particular, severe radiation retinopathy is dose dependent with respect to both total dose (>25 Gy) and dose per fraction (>2 Gy).104,105 Muller et al.103 conducted a prospective study on 102 patients with uveal melanoma treated with fractionated SRT between 1999 and 2007. Patients had uveal melanoma of the choroid or ciliary body with a tumor thickness <12 mm and diameter <16 mm with no metastases. The technique, like GKR, utilized a fixed immobilization system, and a dose of 50 Gy was delivered in 5 equal fractions on 5 consecutive days using 6 MV photons with stereotactic arcs. With median follow-up of 32 months, local control was achieved in 96% of patients. Fifteen enucleations were performed 2 to 85 months after radiation, and best corrected visual acuity (defined as 20/é while using glasses if needed) decreased from a mean of 0.26 at diagnosis to 0.16, 3 months after radiation and then declined to 0.03, 4 years after therapy. Deterioration of visual acuity was in part related to complications of treatment. Grade 3 or 4 neurovascular glaucoma occurred in 9 patients, 8 of which required enucleation. In these 9 patients, tumors were not anteriorly located nor did they receive a high dose to the ciliary body, as one might expect, but were associated with grade 3 retinopathy, which was seen in 19 patients. The authors hypothesized that the physical reaction of ischemic retinopathy might also affect vessels in the ciliary body. Thirteen patients (13%) developed grade 3 or 4 optic neuropathy, which was associated with posterior tumors and optic nerve dose, which was limited to 4 Gy per fraction. Grade 3 cataracts occurred in 10 patients (10%), which were managed by extraction and lens implantation. Cataract formation was dose related, with a median dose of 5 Gy per fraction to the lens, causing cataract in 50% of cases. The authors concluded that while local control was excellent, the number of secondary enucleations was substantial, particularly from neurovascular glaucoma. These data suggest that further follow-up is needed to determine response durability and late side effects of this treatment program.
Retinoblastoma
Retinoblastoma (Rb), the most common ocular malignancy in childhood, affects approximately 300 children per year in the United States.104 The incidence is higher in developing countries, and while the reason for this is not clear, lower socioeconomic status and the presence of certain human papilloma virus sequences have been implicated.105 Rb has a heritable form and a nonheritable form, with approximately 55% of children having the nonheritable form. If there is no family history, the disease is labeled sporadic, but this does not necessarily indicate that it is the nonheritable form, because bilateral cases, most of which are heritable, often have no family history of Rb. It can present with unilateral disease (two-thirds of cases), bilateral disease, or rarely with tumor in both eyes and the pineal gland, which is called trilateral disease.106 Approximately 80% of children in Rb are diagnosed before the age of 3, with unilateral cases diagnosed at an earlier age (14–16 months) than those with bilateral presentations (29–30 months).107–110 Histologically, Rb develops from immature retinal cells and replaces the retina and other intraocular tissues. Macroscopically, viable tumor cells are found near blood vessels, while zones of necrosis are found in relatively avascular areas. Microscopically, both undifferentiated and differentiated elements may be present. Undifferentiated elements appear as collections of small, round cells with hyperchromatic nuclei; differentiated elements include Flexner-Wintersteiner rosettes, Homer-Wright rosettes, and fluerettes from photoreceptor differentiation.111
The study of Rb has provided insights into the genetic basis of cancer. The Rb gene (RB1) is located on the long arm of chromosome 13 (13q14). In order for Rb to develop, both copies of the gene at the 13q14 locus must be lost, deleted, mutated, or inactivated. If either the maternal or paternal copy of the gene that is inherited by an individual is defective, then that individual is heterozygous for the mutant allele. Tumor formation requires both alleles of the gene to be mutant or inactive. These two mutations correlate to the two “hits” theorized by Knudson112 and Hethcote and Knudson,113 which was based on the finding that children with bilateral Rb developed multifocal, bilateral tumors at an earlier age than those with unifocal, unilateral tumors. The first “hit” can be inherited and would be present in all cells in the body, and the second “hit” results in loss of the remaining normal allele and occurs within a particular retinal cell or cells with dysregulation of the cell cycle.114 In sporadic, nonheritable Rb, both hits occur within a single retinal cell after fertilization (somatic events), thus resulting in unilateral Rb. Identifying the RB1 mutation can have management implications both in the affected child as well as siblings and future offspring. For example, if RB1 mutation is detected, then siblings, children, and other relatives can be tested for the mutation. If they do not carry the mutation, they need not undergo rigorous examinations under anesthesia.115
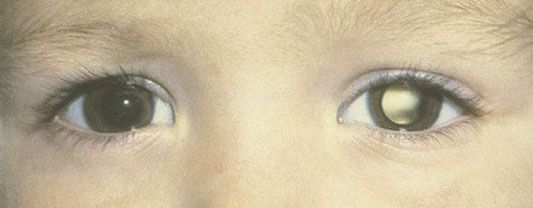
The most common and obvious sign of Rb is leukokoria, a discoloration of the pupil (Fig. 38.7). Other less common and less specific signs and symptoms are deterioration of vision, a red or irritated eye, faltering growth, or developmental delay.116 Some children with retinoblastoma can develop a squint, commonly referred to as cross-eyed or wall-eyed, indicating strabismus.117 In advanced disease in developing countries, eye enlargement is a common finding. Funduscopy typically reveals a white-colored main tumor (Fig. 38.8), frequently with satellite lesions in the retina, subretinal space, or vitreous referred to as “seeds.” Secondary serous retinal detachment may be associated with large lesions. To confirm these findings, a detailed examination under anesthesia through dilated pupils is performed.
Ultrasonography of the eyes is often performed to evaluate the intraocular mass with attention to heterogeneity and calcifications, which support a diagnosis of Rb. Ultrasonography is not as sensitive as CT, which is the ideal imaging format to detect intraocular calcifications. CT, however, raises the concern of exposure to radiation in children younger than 1 year of age with germline mutations,118 but it is still frequently used to confirm the diagnosis. Magnetic resonance imaging (MRI) of the brain and orbits is the most sensitive means of evaluating for extraocular extension and also provides better delineation of the optic nerve and the pineal area.119 MRI of the brain and spinal cord and cerebral spinal fluid examination are indicated when there is gross invasion of the optic nerve by imaging studies or microscopic involvement beyond the lamina cribrosa on histopathologic examination of the enucleated eye. A bone marrow examination and a bone scan are indicated only in cases of an abnormal blood count or clinical symptoms suggesting osseous metastases. The diagnosis of retinoblastoma is based on examination by an ophthalmologist and imaging studies. Biopsy is generally not performed due to the theoretical risk for extraocular dissemination, which could convert an intraocular, curable tumor into extraocular, metastatic disease. Therefore, in the absence of a tissue diagnosis, benign conditions that can resemble retinoblastoma must be carefully excluded.
Rb can spread in a variety of ways, including direct invasion of the optic nerve into the chiasm or dissemination through the subarachnoid space to the brain and spinal cord. Tumors can also invade the choroid and the vascular layer and spread via blood to the bone and bone marrow.120,121 Anterior spread can occur and involve the aqueous venous channels, conjunctiva, and lymphatics or invade the sclera into the orbit with eventual spread to regional lymph nodes.
FIGURE 38.7. Leukokoria from retinoblastoma. (From Aerts I, Lumbroso-Le Rouic L, Gauthier-Villars M, et al. Retinoblastoma. Orphanet J Rare Dis 2006;1:31.)