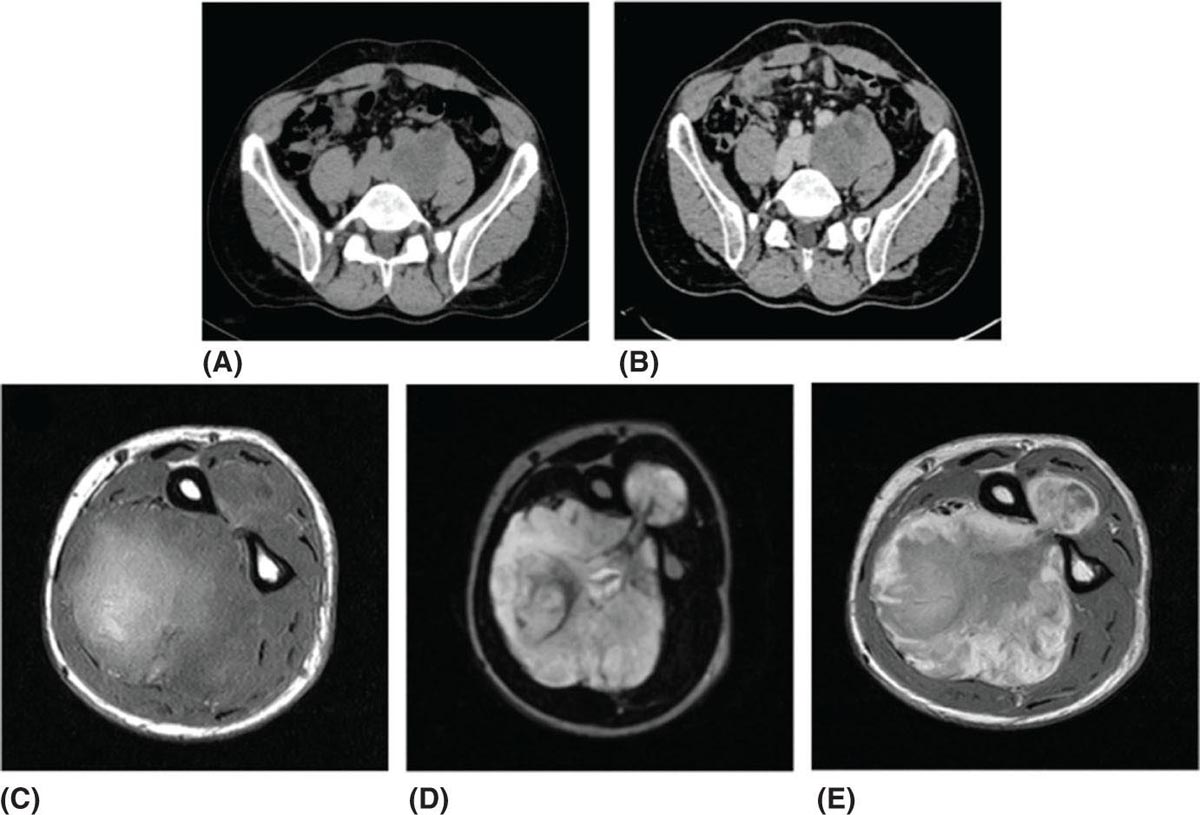
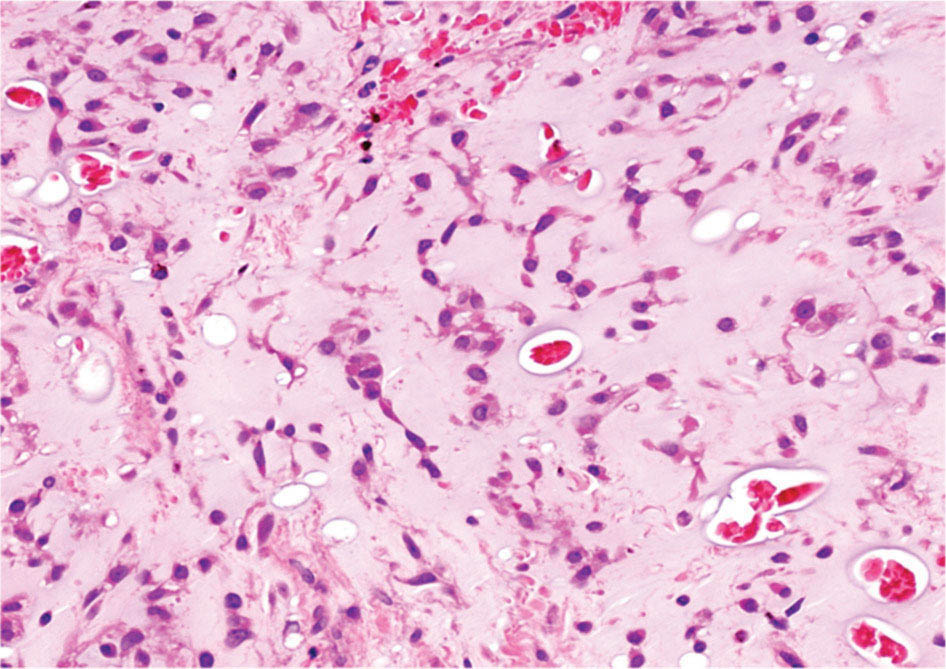
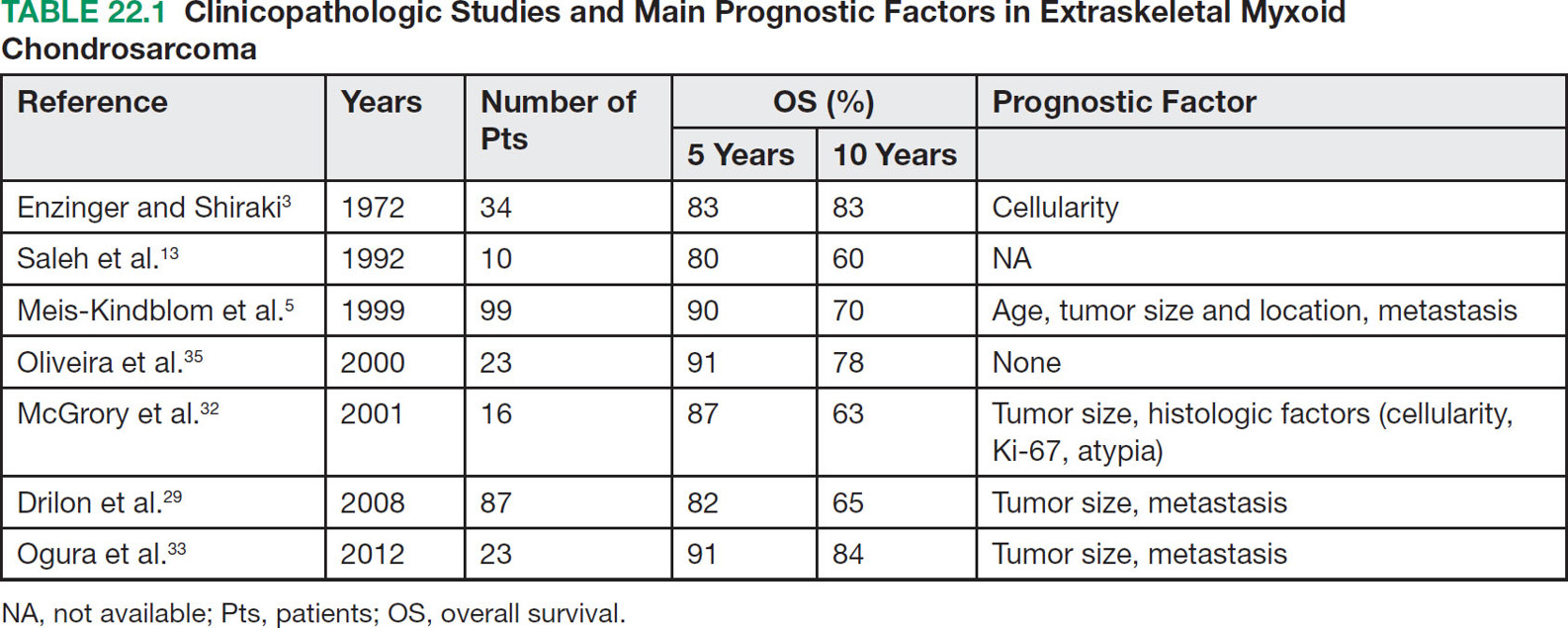
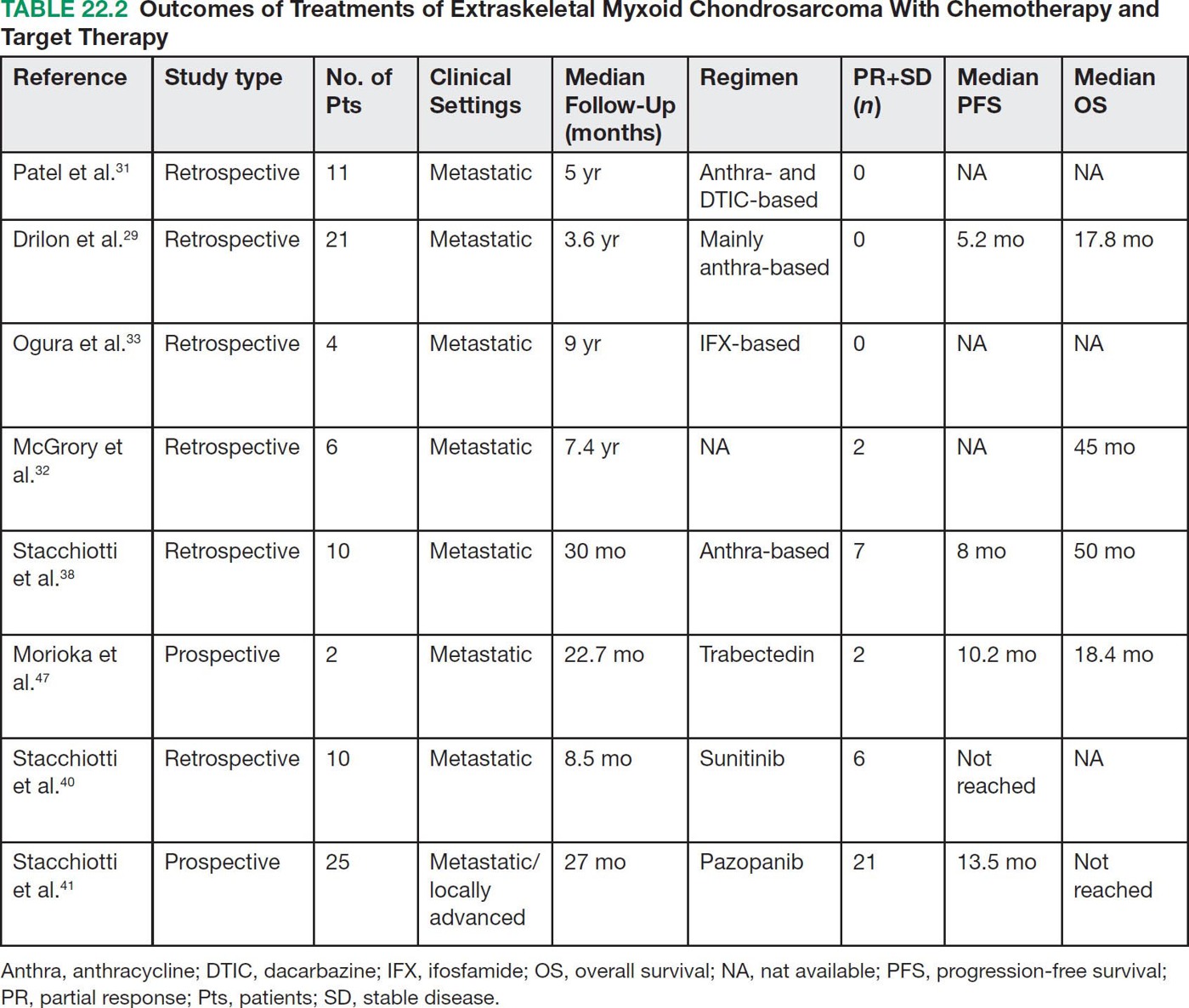
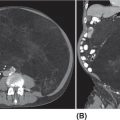
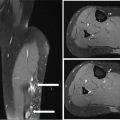
28922 Extraskeletal Myxoid Chondrosarcoma Extraskeletal myxoid chondrosarcoma (EMC) is one of the rarest sarcoma subtypes, mostly arising from soft tissue and rarely from bone. Despite its rarity, the radiographic features, morphology, immunohistochemistry, and molecular biology of the disease are well known and make a reliable diagnosis possible. The natural history, prognostic factors, role of adjuvant/neoadjuvant chemotherapy and radiotherapy in localized cases, and optimal sequence of medical treatments in the advanced phase and patterns of response are not yet well defined, so further research efforts are needed in this disease. Several promising new drugs and new therapeutic strategies are currently under study within ongoing clinical trials that address specific subtypes leading to more personalization of sarcoma treatment. This chapter focuses on diagnostic approach, pathology, and general therapeutic approach for EMC, and includes a discussion of therapeutic approach for metastatic disease. extraskeletal myxoid chondrosarcoma, translocation related sarcomas, EWSR1-NR4A3 genes fusion, antiangiogenics therapy chemotherapy, clinical trials, extraskeletal myxoid chondrosarcoma, history, prognostic factors, quality of care, radiotherapy Chondrosarcoma, Clinical Trials as Topic, Drug Therapy, History, Prognosis, Quality of Health Care, Radiotherapy INTRODUCTION AND EPIDEMIOLOGY Extraskeletal myxoid chondrosarcoma (EMC) is an exceedingly rare soft tissue sarcoma (STS) accounting for <3% of all STSs. It usually arises in adults with a peak incidence in the fifth decade and has a a male predominance (M/F ratio 2:1), with few cases described in childhood and adolescence.1 EMC is a morphologically distinctive neoplasm, described for the first time in 1953 by Stout and Verner2 under the name of chondrosarcoma of the extraskeletal soft tissue and formally defined as a distinctive entity under the name of EMC by Enzinger and Shiraki in 1972.3 EMC is classified by World Health Organization (WHO) as a tumor of uncertain differentiation.1,4 In fact, despite the name, there is no convincing evidence of cartilaginous differentiation, with no clinicopathologic or genetic features in common with any chondrosarcoma subtype. Some molecular studies had shown neuroendocrine properties, raising the possibilities of multiphenotypic differentiation.5,6 EMC arises in deep soft tissues of the proximal extremities and limb girdle in approximately 80% of cases, thigh being the most common site and accounting for 30% to 40% of all cases. Less commonly involved anatomic locations include the head and neck region, the paraspinal soft tissue, the trunk (mostly chest wall and abdominal wall), and the pelvis.4 Pediatric cases of EMC seem to have the propensity to occur in uncommon locations such as scalp, parotid gland, urethra, and sacrococcygeal region. Exceptionally, EMC can arise primarily in bone.7–9 Clinically, EMC usually presents as an enlarging, deep-seated soft tissue mass, often accompanied by pain and tenderness; in some cases, EMC can mimic a hematoma, and when arising around joints, it can produce functional impairment. DIAGNOSTIC APPROACH The recommended imaging of the primary tumor is MRI. Because lymph node involvement is more frequent than observed in the other STSs, regional lymph node status assessment through CT/MRI is advisable. Radiographically, because of the presence of a rich myxoid matrix, EMC usually appears as a low attenuation lesion on CT scan and displays hyperintense signal with hypointense internal septa on T2-weighted MRI, which is reflective of a high water content. They also have pronounced lobular architecture often invading extracompartmental, bony, and vascular structures. As the tumor may undergo hemorrhagic and necrotic degeneration, signal characteristics on T1-weighted MRI can vary, ranging from low-, intermediate- and high-intensity features compared to muscle.10–12 Peripheral/septal enhancement was observed in nearly one-third of the cases after the administration of contrast medium. In addition, serpentine high-flow vessels may be seen: they correspond to the hemangiopericytoma-like areas, which can be identified histologically (see Figure 22.1). Similar to other STSs, a chest and abdomen CT scan is mandatory for staging purposes; metastases are usually pulmonary, but extrapulmonary metastases can less commonly occur. Fluorodeoxyglucose (FDG)-PET/CT imaging may be useful in selected cases to investigate tumor aggressiveness.11 As for all sarcomas, pathologic diagnosis is mandatory. Usually a core-needle biopsy taken within a sarcoma reference center is considered the best approach. FIGURE 22.1 (A–B) Primary retroperitoneal extraskeletal myxoid chondrosarcoma (EMC) on axial pre- and postcontrast administration on CT scan. (C) Primary left forearm EMC on T1-weighted MRI shows a focus of high signal intensity within the tumor resulting from hemorrhage. (D) Primary left forearm extraskeletal myxoid chondrosarcoma on T2-weighted MRI shows hyperintense signaling with hypointense internal septa. (E) Primary left forearm extraskeletal myxoid chondrosarcoma on T1-weighted MRI after contrast administration with typical peripheral/septal enhancement. PATHOLOGY AND MOLECULAR BIOLOGY Microscopically, EMC typically features multinodular architecture defined by fibrous septa that divide the tumor into hypocellular lobules with abundant pale blue myxoid or chondromyxoid matrix.1 The stroma is strikingly hypovascular. The cells characteristically grow in cords, strands, and even more complex trabecular or cribriform arrays (Figure 22.2). Cytoplasm is eosinophilic to vacuolated and harbors uniform round to oval nuclei. Mitotic activity is usually low. Some tumors have prominent rhabdoid cytoplasmic inclusions. Rare cases are hypercellular with decreased myxoid matrix and have higher grade, often epithelioid cytomorphology.13,14 Well-formed hyaline cartilage is virtually never seen. FIGURE 22.2 Extraskeletal myxoid chondrosarcoma neoplastic cells typically grow in cords set in abundant chondromyxoid matrix (hematoxylin & eosin stain). From the molecular standpoint, EMC is characterized by the rearrangement of the NR4A3 gene, also known as NOR1. NR4A3 gene rearrangement represents an important tool to enable differential 291diagnosis with other mimics, in particular myoepithelial neoplasms. NR4A3 has been implicated in regulating key cellular functions, including proliferation, cell survival, and inflammation.15–17 In most cases, the NR4A3 gene is rearranged with the EWSR1 gene (approximately 70% of cases) as a result of a t(9;22)(q22;q12) translocation. Less frequently, NR4A3 has been found rearranged with other genes and in particular TAF15 (17q12), more rarely TCF12 (15q21), TGF (3q12.2), or FUS (16p11.2). The spectrum of NR4A3 rearrangements is, however, evolving in EMC, and recently HSPA8 was also identified as a novel fusion partner of NR4A3.18–21 While the EWSR1-NR4A3 and TAF15-NR4A3 fusion proteins are strong transcriptional activators, the HSPA8-NR4A3 could act as a transcriptional repressor,22 assuming different pathogenetic mechanisms of the disease based on the presence of different translocations. A clear correlation between NR4A3 gene fusion variants with morphology and clinical outcome has not been identified to date, though retrospective studies showed that TAF15-NR4A3 subtype had more aggressive features. In particular, TAF15-NR4A3 positive EMC showed a higher incidence of rhabdoid phenotype, high-grade morphology (solid pattern of growth, marked nuclear atypia, high mitotic activity), and a more aggressive clinical outcome compared to the EWSR1–NR4A3 cases.23 Recently to highlight this issue and to provide grounds for better risk stratification criteria, a set of EMC samples and cell models expressing either EWSR1 or the TAF15 fusion transcript were molecularly profiled by RNA-seq showing that the two EMC molecular variants display a distinct transcriptional profile with axon guidance and neurogenesis pathways as major discriminants. In particular, class 4 to 6 semaphorins with a pro-tumorigenic activity were more often expressed in TAF15-NR4A3 tumors, while class 3 semaphorins that convey growth inhibitory signals were more abundant in EWSR1-NR4A3 tumors. These results suggest that the type of NR4A3 fusion could dictate an axon guidance switch and impact on tumor cell biology.24 Finally, deletions or mutations involving the SMARCB1 gene have been identified in a small subset of EMC, particularly in cases with rhabdoid morphology.25,26 GENERAL THERAPEUTIC APPROACH Currently the only curative option for EMC, when feasible, is wide local resection with negative margins (remove the tumor with a rim of normal tissue around it), that is the standard surgical procedure as for the all patients with an adult type, localized STS.27,28 A surgeon specifically trained in the treatment of this rare disease must carry it out. The use of radiation therapy (RT) for localized disease in the adjuvant or neoadjuvant setting is still debated, because the impact of RT has largely gone understudied in this histotype, and historically, it has been considered a chemotherapy- and RT-resistant tumor. A retrospective study of 87 patients with EMC treated at two referral institutions between 1975 and 2008 showed a high rate of local recurrence in patients who underwent surgical resection without RT (35% at a median follow-up of 2.2 years).29 Recently, a propensity score weighted, population-based analysis of the Surveillance, Epidemiology and End Results (SEER) database was performed to examine the effect of external beam RT (EBRT) on cancer-specific survival for 156 patients with localized EMC as first malignancy.30 There were 147 patients (94%) who received surgery and 50 patients (32%) who received EBRT. Forty-five patients (29%) received both surgical resection and EBRT. At a median follow-up of 33 months, the authors observed a cancer-specific survival rate of 97% versus 85% and 94% versus 85% in favor of EBRT at 3 and 5 years, respectively. In this analysis tumor size, locoregional lymph node involvement, and type of surgery significantly correlated with cancer-specific survival. Based on these results, we could extrapolate that therapeutic strategies that include aggressive local treatment with surgical resection and EBRT should be considered in localized disease, in particular for surgically removed large lesions and/or cases with locoregional nodal involvement. Neoadjuvant RT is preferable when tumor shrinkage is needed for a surgical downstaging when function preservation is the goal. By contrast, there are no data to support the use of adjuvant/neoadjuvant chemotherapy in the disease unless high-grade features are seen.27,29,31–33 In this case, chemotherapy is still not standard, but may be offered as an option for shared decision-making with the patient.34 In our experience, some patients with locally advanced and marginally resectable disease treated with chemotherapy and RT, also in combination in the preoperative setting, experienced significant responses, despite this tumor being commonly believed to be resistant to chemotherapy and RT; this kind of response allowed us in some cases to perform less extensive surgery than originally planned, especially for critical anatomic sites. PROGNOSTIC FACTORS AND PROPENSITY TO METASTASIZE EMC have usually an indolent natural history, but a more aggressive course is observed in high-grade and/or dedifferentiated cases. The rate of both local recurrence (range from 35% to 50%) and distant metastasis (range from 25% to 50%) is high in EMC, although metastases often occur late after the diagnosis of primary tumor (Table 22.1)2,3,13,14,27,29,35 Ten-year survival ranges between 85% and 65% and 5-year survival is approximately 90%.29,33 As already discussed, the most common sites of metastasis are the lung, lymph nodes, bone, and other soft tissue sites. Based on the few data available, older age, proximal extremity location, larger tumor size (>10 cm), and high-grade confer a worse prognosis.4,32 There are few published data to indicate the optimal routine follow-up policy for surgically treated STS patients with localized disease36 and in particular for EMC patients. Given the risk of local recurrence and distant metastasis, regular follow-up after excision of primary disease, especially for the high-grade variant of EMC, is advisable. The recommended follow-up should include a CT scan of thorax and abdomen and an MRI of the primary tumor site. Because metastatic disease can occur late in EMC, with more than 70% of local recurrences and first metastases of EMC happening beyond 5 years, longer follow-up is suggested. THERAPEUTIC APPROACH FOR METASTATIC DISEASE As for all STS, surgical resection can be a reasonable option with curative intent, taking into accont the expected morbidity in cases of limited, resectable, slow growing lung metastases.37 By contrast, patients with advanced disease not amenable to surgical resection cannot be cured and may need medical treatment, expecially with evidence of disease progression. Data available in the literature on the activity of anticancer agents in EMC are few and mostly retrospective. While retrospective series published some years ago reported very limited activity of chemotherapeutic agents generally used for STS (for example, anthracycline-based, dacarbazine-based or ifosfamide-based regimens) (Table 22.2),29,31–33 more recently, a retrospective study on a small series of molecularly confirmed EMC patients treated with anthracycline-based chemotherapy,38 showed better results. In this study, four partial responses by the Response Evaluation Criteria in Solid Tumors (RECIST) of 10 evaluable patients, with a median progression-free survival (PFS) of 8 months were described. Although to be confirmed prospectively and/or on larger series, a possible explanation for the different results observed in this study compared to the previous ones is the selection of cases with a diagnosis confirmed by the presence of the NR3A4 fusion. Other medical options include antiangiogenic agents. The first evidence on the activity of an antiangiogenic agent in EMC was reported in 2012, and consisted of a RECIST partial response39 observed to sunitinib in two patients with an NR4A3-EWS positive, progressive metastatic EMC. Afterward, sunitinib activity in the same patient population was confirmed in a retrospective series of 10 patients,40 with six of 10 partial responses and with median PFS not reached at a median follow-up of 8.5 months. Curiously, all responsive cases were EWSR1-NR4A3 positive while the 2/10 cases carrying the TAF15-NR4A3 fusion progressed.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


