Ewing Tumor
 EPIDEMIOLOGY
EPIDEMIOLOGY
Ewing sarcoma family tumor (ESFT) is the second most common primary tumor of bone in childhood, and also arises in soft tissues. ESFT is uncommon before 8 years of age and after 25 years of age.1 In the European Intergroup Cooperative Ewing Sarcoma Study group (EICESS), the median age was 14 years, with 57% of the patients male and 43% female, although a majority of the patients below 10 years of age were female.1 The malignancy is rare in African Americans.2 In the EICESS, 24.7% of lesions are located in the pelvis, 16.4% in the femur, 16.7% below the knee, 12.1% in the ribs, 8.0% in the spine, and 4.8% in the humerus.1
 PATHOLOGY AND CYTOGENETICS
PATHOLOGY AND CYTOGENETICS
Light microscopy shows a tumor of small, round blue cells that lack markers for lymphoma, neuroblastoma, or rhabdomyosarcoma. Cytogenetics has shown that Ewing tumors of bone and soft tissue are the most undifferentiated members of a tumor family that shares a common neuroectodermal precursor cell, arrested at different stages of differentiation.3,4 Approximately 95% of ESFTs have a translocation between the EWS gene on chromosome 22 and the FLI1 gene on chromosome 11 (t[11;22][q24;q12]) or the ERG gene on chromosome 21 (t[21;22][q22;q12]),5,6 although in an analysis of 222 consecutive tumors at the Rizzoli Institute in Italy with a presumptive diagnosis of Ewing tumor, an occasional other translocation was noted.7 The translocations are present only in tumor cells and occur in bone and soft-tissue Ewing tumors, primitive neuroectodermal tumors of bone and soft tissue, peripheral primitive neuroectodermal tumors, Askin tumors, some esthesioneuroblastomas in children, and some central nervous system (CNS) tumors.8 Intra-abdominal desmoplastic small round cell tumor appears to have a different translocation at t(11;22)(p13;q12), indicating it is not one of these neoplasms.9 The proto-oncogene c-myc, not seen in neuroblastoma, is expressed in the ESFT, whereas n-myc is not amplified.9
 CLINICAL PRESENTATION
CLINICAL PRESENTATION
The most frequent presenting symptoms are pain and swelling. Pain can wax and wane as the tumor progresses. Symptoms of systemic disease occur at times, including low-grade fevers, malaise, and weakness. ESFT patients exhibit a mean lag time of 146 days between the onset of symptoms and diagnosis, the longest lag time of any pediatric solid tumor.10 Both patients and physicians contribute to this delay. Overall, patients with the EWS-FLI1 fusion have a similar clinical presentation and prognosis to those with the less common EWS-ERG fusion.11
 DIAGNOSTIC WORKUP
DIAGNOSTIC WORKUP
In Ewing tumor of bone, plain films usually reveal a mottled or moth-eaten lesion. Lytic and blastic areas may be present; lytic areas are more commonly seen. Subperiosteal reactive new bone may be present, producing an “onion skin” appearance. Like an osteosarcoma, Ewing tumors may produce spicules radiating from the cortex of the involved bone, or expansion of the bone may produce a cystic-appearing tumor. Occasionally, the tumor may appear to arise on the surface of the bone, producing a saucerlike indentation of the surface.12 A magnetic resonance imaging (MRI) scan of the primary lesion will show the extent of bone marrow involvement and soft-tissue invasion, whereas bone destruction is best seen using computed tomography (CT). The radiographic differential includes osteosarcoma, osteomyelitis, eosinophilic granuloma, primary lymphoma of the bone, and even an occasional metastatic malignancy.
The systemic workup should include blood studies, a chest roentgenogram, a CT scan of the chest, a bone scan, and a bone marrow biopsy. Fluorodeoxyglucose (FDG)-positron emission tomography (PET) scans have been shown to detect considerably more bone metastases than traditional bone scans, both at diagnosis and recurrence. Whole-body MRI has also been reported to be superior to bone scan in detecting bone metastases in patients with Ewing tumors, particularly diffusion-weighted images or short-time inversion recovery (STIR) images, but some lesions are found on bone scans that are not seen on MRI.13,14 MRI has also been reported as better than FDG-PET.15 Because few lesions <8 mm are detectable using PET imaging, CT scans are still more accurate for the screening of lung metastases.16 Nevertheless, routine CT scans of the abdomen and pelvis rarely pick up metastatic disease not shown on other studies, either in the initial workup or during follow-up.17
In a large cooperative group study, approximately 20% of patients presented with metastatic disease. Of these patients, 44% presented with lung metastases only, 51% with bone or bone marrow involvement (with or without lung metastases), and 5% with metastases in other organs.1 However, three times as many bone lesions are diagnosed in studies utilizing whole-body MRI scans,13,14 so it is quite possible that the standard workup using only bone scans underestimates the incidence of bone metastases. Because of the specific cytogenetic identity of ESFTs, cytogenetic analyses have been shown to occasionally diagnose a Ewing tumor in the setting of an unknown primary tumor.18
 PROGNOSTIC FEATURES
PROGNOSTIC FEATURES
Metastases at diagnosis, a large or pelvic or truncal primary tumor, the presence of a large soft-tissue mass, an older age at diagnosis, a poor response to induction chemotherapy, not using surgery as part of the treatment of the primary lesion, and a filigree histologic pattern have all been proposed as poor prognostic factors.1,19,20,21,22,23,24,25,26,27 These factors are interrelated; 90% of tumors in the pelvis and femur are usually large, have a large soft-tissue mass, and occur more often in older adolescents and adults.28 The presence of necrosis on pretreatment MRI scans has been linked to an increased risk of metastases at diagnosis.29
There are differing opinions regarding the prognosis of skeletal versus nonskeletal primary lesions. Some reports show an advantage for skeletal primary sites,30 but if size, site, and other tumor characteristics are stratified, the prognosis may be identical.31
The radiologic or histologic response of the soft-tissue mass to induction chemotherapy is an extremely good prognostic indicator1,24,27,32; the PET scan response is different from osteosarcoma and not as useful.33
Ewing tumors with p53, p16/p14ARF alterations or the presence of vascular endothelial growth factor respond poorly to chemotherapy and have a poor prognosis.34,35,36 The matricellular protein CCN3, which plays an important role in bone formation, has been reported to be associated with a worse prognosis if fully expressed.36
Although in the past the type 1 fusion abnormality, which is the EWS-FLI1 transcript created as a result of fusion between exons 7 of EWS and 6 of FLI1, has been reported to have a favorable prognosis, more recent analyses do not confirm this. A re-examination of this association in a prospective cohort of patients with ESFT treated according to current Children’s Oncology Group (COG) protocols using more intensive chemotherapy show no advantage.37 In a report from the Euro-Ewing 99 trial, no type of translocation impacted disease progression or relapse.38
The EWS-FLI1 transcript can sometimes be detected in the peripheral blood or bone marrow, where it may indicate residual occult disease.39
 GENERAL MANAGEMENT
GENERAL MANAGEMENT
Effective local and systemic therapy is necessary for the cure of ESFTs. Most chemotherapy regimens combine cyclophosphamide, doxorubicin HCl (Adriamycin), vincristine, dactinomycin, ifosfamide, and etoposide.21,23,27,40,41,42,43,44 Induction chemotherapy is preferred over starting the systemic therapy and local therapy concomitantly. There are several advantages to this approach:
1. Administering the chemotherapy first allows an evaluation of the effectiveness of the regimen for each patient;
2. Shrinkage of the soft-tissue mass may help the surgeon or radiation oncologist decrease the volume of the local therapy;
3. Shrinkage of the soft-tissue mass may allow the surgeon to achieve better margins; and
4. Some bone healing takes place during the chemotherapy, which may diminish the risk of pathologic fracture if radiation therapy is used to treat the primary lesion.
Response rates to induction chemotherapy are high, with radiologic complete response and partial response rates of up to 90% reported.32,45,46 For institutions that use surgery for the treatment of the primary lesion, excellent necrosis rates have been reported in many patients.19,27 Almost all patients whose lesions show a poor response either radiologically or histologically die.19,24,27,32
The biopsy should be performed at the same institution where the treatment will be performed, and the biopsy specimen should only be taken from the soft-tissue component, if present. Enough tissue should be collected for light and electron microscopy as well as cytogenetics. In experienced hands, a large-needle biopsy may be sufficient, although usually a larger sample is preferred, particularly because cytogenetics is becoming increasingly important.
For definitive therapy, limb-salvage surgery is preferable over amputation, but amputation may be an option for younger patients with lesions of the fibula, tibia, and foot. In older patients, lesions of the proximal fibula, ribs, scapula, clavicle, and wing of the ilium are easier to resect than other sites. Lesions of the bones of the hands and feet may be resectable with a ray resection. Other sites may be resectable with major reconstructive procedures and significant morbidity.47
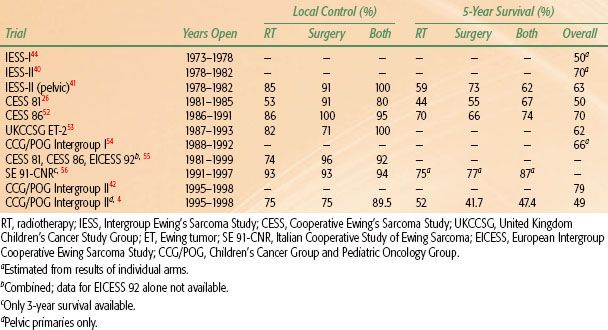
TABLE 88.1 SURVIVAL AND LOCAL CONTROL BY METHOD OF TREATMENT TO PRIMARY LESIONS IN MAJOR COOPERATIVE GROUP TRIALS (LOCALIZED LESIONS ONLY)
 RADIOTHERAPY TECHNIQUES
RADIOTHERAPY TECHNIQUES
For gross disease, standard treatment is a total dose of 55.8 Gy at 1.8 Gy per day, with a field reduction at 45 Gy, although 36 Gy may be adequate for the initial field.48,49,50,51 Local control rates of 53% to 93% have been reported with these doses (Table 88.1). Local control at doses <40 Gy is significantly worse, even for small lesions.57
Twice-a-day irradiation has been used in several trials. To treat ESFTs, radiation oncologists at the University of Florida used 1.2 Gy twice a day to a total dose of 50.4 Gy, 55.2 Gy, or 60 Gy, depending on the tumor response to induction chemotherapy, and showed that late effects could be decreased while maintaining good local control, even for large primary tumors.48,51 In CESS 86, doses of 1.6 Gy twice a day to a total of 60 Gy were given, although not in a continuous course. The Italian SE-91 trial also used 1.6 Gy twice a day to a total of 60.8 Gy, but in a continuous course. The CESS 86 trial showed no advantage for the accelerated hyperfractionated approach, but the early results of SE-91 are promising with regard to local control.52,56 Both of the latter regimens would be theoretically expected to increase late effects.
The Pediatric Oncology Group trial (POG 8346) showed, in a randomized fashion, that the traditional approach of irradiating the entire marrow cavity was not necessary.58 Although “tailored” fields are the standard of care today, attention to the requisite volume is critical to obtaining maximal control rates; geographic miss has been a frequent source of failure in cooperative group studies.26,58 Three-dimensional treatment planning is essential. In the present open COG trial, the initial clinical target volume 1 includes the prechemotherapy tumor volume with a 1-cm margin, with at least a 0.5-cm addition for planned target volume 1. The boost is to the residual tumor volume at the time of radiotherapy plus a 1-cm margin, with or without a 0.5-cm planned target volume 2. PET-CT may be helpful for planning. Any initial bone or bone marrow abnormalities in the primary bone should be included in gross tumor volume 2. These margins are much tighter than in previous studies, and only additional follow-up will show whether they will be adequate.
Extremity lesions require sparing at least a 1- to 2-cm strip of tissue to prevent lymphedema, which can be very difficult at times, particularly in arm lesions, although the arm is less likely to develop lymphedema than the leg. It may be necessary to consider surgery as an alternative if a strip of tissue cannot be spared. It is more important to cover the tumor adequately than to spare adjacent growth plates and joints.
With the high doses of cyclophosphamide or ifosfamide given in chemotherapy regimens for Ewing tumor, it is important to minimize the dose to the bladder. Radiation cystitis can be a significant risk even at doses as low as 20 Gy. Because pelvic lesions rarely infiltrate into the tissues around the bladder, but instead tend to push aside those structures, neoadjuvant chemotherapy allows additional bladder to be spared if good shrinkage is obtained. A 1-cm medial margin on the residual disease at the time of treatment is adequate from the beginning of radiation therapy.
Rib lesions should be treated conformally with a minimum of lung and heart in the high-dose field. However, rib primary tumors often present with pleural effusions, and in the EICESS and COG trials hemithorax irradiation was used, even for surgically resected lesions. A dose of 15 Gy for patients younger than 14 years and 20 Gy for older patients was given, corrected for lung transmission. In the EICESS 92 trial, the 7-year rate of event-free survival (EFS) was 63% with hemithorax irradiation versus 46% without.59 Another option is 32P colloid, which has been used in addition to standard radiation, but not hemithorax, in high-risk but localized chest wall tumors in an attempt to decrease chest or pleural recurrence, with no relapses in the patients receiving it.60
In the United States, the standard dose for vertebral lesions is 45 Gy; it is not clear whether this decreases local control and survival rates. The average dose used in the CESS 81, CESS 86, and EICESS 92 trials was 49.6 Gy, although sometimes spinal shielding was used.61,62 In these trials, neurologic late effects were seen in one of 47 patients irradiated, occurring at a dose of 44.8 Gy at 1.6 Gy twice a day. Sacral lesions should be treated to the full dose.
Doxorubicin and dactinomycin given during the course of radiation therapy will often cause moist desquamation, particularly with beams of 6 MV or less, tangential irradiation, or skin folds. These drugs may also cause a “recall” phenomenon of the dry or moist desquamation when given after the end of radiation therapy.
The indications for adjuvant radiotherapy with surgery are not completely defined. Jereb et al.63 found that the local recurrence rate after conservative surgery was high without irradiation. Ozaki et al.64 also reported a slight advantage for adding postoperative irradiation for patients with inadequate margins. Dunst and Schuck65 reported that patients with a wide resection alone but a poor histologic response had a local failure rate of 12%, in comparison with 6% for similar patients who received postoperative radiotherapy. With present data, postoperative radiotherapy should probably be given to all patients with marginal margins and all patients with a poor histologic response. Intralesional surgery is not indicated as the EICESS trials showed no improvement in local control in patients with intralesional surgery plus radiotherapy versus radiotherapy alone.65
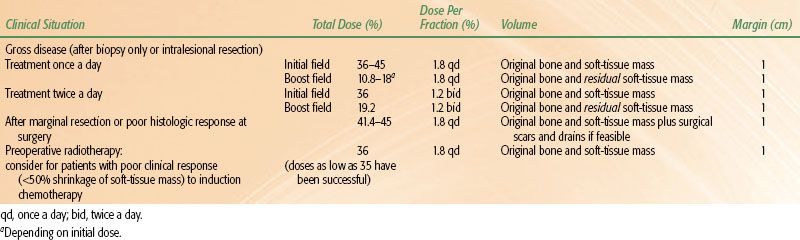
In the COG trials, a dose of 50.4 Gy at 1.8 Gy once a day is given if postoperative radiotherapy is indicated. Doses in the range of 30 to 44.8 Gy at 1.8 Gy a day have also been reported to be effective for subclinical disease.64,66 Intralesional resections should be treated to the same dose as in patients receiving radiotherapy alone. Table 88.2 lists the recommended doses and fields for different clinical situations.
In a review of 153 patients treated with surgery followed by postoperative radiotherapy from CESS 86 and EICESS 92, Schuck et al.67 showed that the interval between surgery and radiotherapy did not influence survival, although there was a slight trend for improved local control in patients receiving radiotherapy <90 days postoperatively.
Good results have also been reported with preoperative radiotherapy for patients with a poor response (<50% reduction of the evaluable soft-tissue mass) after two cycles of chemotherapy.61,64 Doses of 36 to 63 Gy have been used.64
Radiation therapy doses and margins are shown in Table 88.2.
TABLE 88.2 RECOMMENDATIONS FOR RADIATION THERAPY FIELDS AND DOSES
 RESULTS OF THERAPY
RESULTS OF THERAPY
The majority of studies reported in the literature reflect the results of therapy only for Ewing tumor of bone, because before 1991 soft-tissue Ewing tumors were treated on Intergroup Rhabdomyosarcoma Study (IRS) Group protocols. The addition of chemotherapy to local therapy increased the survival for Ewing tumor of bone from <10% to >40% at 5 years for patients with localized disease at diagnosis.68 Modern protocols show better results and Surveillance, Epidemiology and End Results (SEER) data confirm this gradual improvement in survival rates over time. Five-year survival improved from 36% for patients treated from 1973 to 1977 to 59% for patients treated from 1993 to 1997.69 However, late recurrences and deaths from complications continue to occur for years, yielding 10- and 15-year survival rates 10% to 15% lower than 5-year rates.32,70
Local control and survival rates are shown in Table 88.1 for patients treated with different local control strategies in cooperative group trials that reported appropriate data. Surgery has become the treatment of choice for the primary lesion at most centers, and many series report superior local control for patients receiving surgery as a component of their local treatment, particularly if radiotherapy compliance was poor.26,58 The influence of different local control strategies on survival is less certain. In CESS 86, with good radiotherapy compliance, survival rates were not influenced by the method of therapy to the primary tumor.52 Nor was there an advantage for surgery in pelvic primary lesions treated in Intergroup 0091.4 Other trials show an advantage for surgery or a combination of surgery and radiotherapy. With no randomized studies addressing this question and considerable selection going into the choice of local treatment, conclusions regarding the relative influence of different local therapies on survival are based more on opinion than fact.26
However, systemic therapy does influence the rate of detectable local relapse. The first Intergroup Ewing’s Sarcoma Study (IESS-I) reported that the addition of doxorubicin improved local control.6 The first Children’s Cancer Group/Pediatric Oncology Group (CCG/POG) intergroup study showed an improvement in survival in the intensified arm for patients with localized disease and large primary or pelvic tumors. The improvement with the more intensive regimen resulted from a decrease in the rate of local relapse; the rate of distant metastases was similar in both arms.42 The time interval between initiation of chemotherapy and start of radiotherapy has also been reported to influence survival (local control was not evaluated), with early radiotherapy preferable to late radiotherapy.65
 RESULTS OF CLINICAL TRIALS FOR PATIENTS WITH LOCALIZED DISEASE AT DIAGNOSIS
RESULTS OF CLINICAL TRIALS FOR PATIENTS WITH LOCALIZED DISEASE AT DIAGNOSIS
IESS-I showed that VACA chemotherapy (vincristine, dactinomycin, cyclophosphamide, and doxorubicin) produced superior 5-year relapse-free survival rates over a VAC (vincristine, dactinomycin, and cyclophosphamide) regimen alone. The third arm combined bilateral whole-lung irradiation (WLI) with VAC and produced survival rates better than VAC alone, but less than VACA, indicating that bilateral WLI is an effective adjuvant, although it has not been studied in any subsequent trial except in patients with lung metastases at diagnosis.44 IESS-I also showed poorer survival rates in all arms for patients with pelvic tumors; intensifying systemic therapy in IESS-II improved the results for pelvic tumors. IESS-II also showed that induction chemotherapy was a viable option.41 A trial at St. Jude Children’s Research Hospital (Memphis, TN) confirmed this approach, and it has since been the standard of care.45
Based on data from previous trials,25 both the University of Florida and the German Cooperative Ewing Sarcoma Study stratified ESFTs by tumor size and intensified treatment for large localized tumors. Although neither approach was randomized, both the University of Florida studies and CESS 86 found that intensifying treatment improved survival closer to the level as achieved with smaller primary lesions and less-aggressive chemotherapy.32,51,71
In the first CCG/POG intergroup study starting in 1988, a randomization between VACA chemotherapy alone versus VACA alternating with etoposide and ifosfamide showed that adding etoposide–ifosfamide improved the survival rates, particularly for patients with primary tumors of the pelvis.42 The United Kingdom Children’s Cancer Study Group trials ET-1 and ET-2 showed a better survival rate in the ET-2 trial if ifosfamide was substituted for most of the cyclophosphamide used in ET-1; however, the dose intensity in ET-2 was also higher.53
The second POG and CCG intergroup trial, accruing 483 eligible patients between 1995 and 1998, built on the previous intergroup protocol. The 48-week vincristine, doxorubicin, cyclophosphamide, ifosfamide, and etoposide regimen was used as the standard arm, compared with the same drugs given in fewer cycles but with higher drug doses per cycle. There was no significant difference in survival rates in the two arms, with a 5-year EFS of 71% and an overall survival of 79%.54
The first COG trial used the more standard doses but decreased the interval of chemotherapy to 2 weeks if there was blood count recovery. The 3-year EFS was 76% for the interval compression arm versus only 63% for the more standard arm.72 Because of the results of the interval compression study, the five-drug standard of vincristine, cyclophosphamide, doxorubicin, ifosfamide, and etoposide with interval compression is now considered the standard Ewing sarcoma regimen in North American institutions. The present phase III COG study is evaluating the addition of the regimen vincristine, topotecan, and cyclophosphamide to the five-drug intensive regimen.
The EICESS 92 trial showed that for standard-risk patients (<100 mL and no metastases at diagnosis), a regimen substituting cyclophosphamide (VACA) for ifosfamide (VAIA) was equivalent.73 In high-risk patients (metastases at diagnosis or primary lesions >100 mL), the addition of etoposide provided superior survival results than VAIA alone.5
Raney et al.74 reported the results of treating extraosseous Ewing tumors on the IRS until 1991. Long-term survival of these patients was probably better than for patients with bony primaries, with 10-year survival of 62%, 61%, and 77% for IRS-I, IRS-II, and IRS-III therapeutic protocols, respectively. Survival rates were better for patients with primary lesions of the head and neck, extremities, and trunk and for those with gross tumor removal. Extraosseous primary lesions are now treated on the same protocol as osseous lesions.
 RESULTS OF CLINICAL TRIALS FOR PATIENTS WITH METASTATIC DISEASE AT DIAGNOSIS
RESULTS OF CLINICAL TRIALS FOR PATIENTS WITH METASTATIC DISEASE AT DIAGNOSIS
Patients with metastatic disease at diagnosis have a poor prognosis. In a report from the EICESS, the 5-year relapse-free survival for patients with lung metastases only was 29%; for patients with bone and bone marrow metastases, it was 19%; and for patients with both lung and bone metastases at diagnosis, it was only 8%.1 Although there was an improvement in survival rates in patients with localized disease treated in the first intergroup CCG/POG trial, adding ifosfamide–etoposide did not improve survival for patients with metastatic disease at diagnosis.75
WLI improved survival in the CESS 81, CESS 86, and EICESS 92 trials for patients with lung metastases as the only site of metastatic disease at diagnosis.76 The CESS 81 and 86 studies also showed increasing survival rates with an increasing radiation dose to the lung fields, particularly with a dose of >18 Gy (corrected for lung transmission) at either 1.5 Gy once a day or 1.25 Gy twice a day. Individual residual lesions were boosted.3 Some institutions only use WLI in patients with lung metastases who respond poorly to chemotherapy.77
In patients with disseminated disease, results are better with adequate treatment of the primary as well as all the metastases. EURO-EWING 99 showed that the 3-year EFS was better (47%) for patients receiving both surgery and radiation to the primary lesion versus either surgery alone (25%) or radiation therapy alone (23%). Without local therapy, the EFS was 13%.78 Obviously, selection bias cannot be excluded. At Memorial Sloan-Kettering Cancer Center (New York, NY), local control was worse for patients with metastatic disease (61%) than those with localized disease (84%),79 although in a large series from St. Jude Children’s Hospital, the local control in the two groups was equal.80
Recent evidence shows improved survival in patients with disseminated bone metastases if all the lesions are treated. Of 120 patients enrolled on arm R3 of EURO-EWING, which suggested but did not mandate local therapy (radiation, surgery, or both) to all sites of disease, 40% received local therapy (either surgery or radiation) or both to all sites of metastatic disease, and 60% received no local therapy. Three-year EFS was 35% in those patients who received radiation therapy to metastatic sites compared to 16% in those who received no local therapy.78 Paulino et al.81 show similar results with local therapy to all sites of metastatic disease. It is acceptable to delay the radiotherapy until close to the end of chemotherapy if a significant amount of bone marrow would be treated.
 HIGH-DOSE THERAPY WITH STEM CELL RESCUE
HIGH-DOSE THERAPY WITH STEM CELL RESCUE
Many institutions have investigated end-intensification with megatherapy and stem cell rescue using high-dose chemotherapy with and without total-body irradiation. Patients with metastatic disease treated on EICESS studies between 1990 and 1995 had a superior 4-year EFS when megatherapy was added to the end of therapy, but CCG-7951 showed no improvement with this approach over a matched cohort from other Ewing tumor studies for patients with bone or bone marrow metastases.76,82 Some institutional pilot studies without strict selection criteria show promising but not conclusive results.32,43,83,84 The use of two sequential transplants (tandem transplants) has produced promising early results.5 Total-body irradiation has been reported to add to toxicity but not improve EFS.5,85
A few institutions have used a single transplant to treat high-risk patients (large primary tumors or tumors of the pelvis or trunk) with localized disease. Five-year survival rates of 48% to 71% have been reported.5,32,43,86 Although the results are clearly better than standard treatment for similar high-risk groups treated historically, recent intensified regimens without end-intensification appear to give similar results,19,32,42,54 showing that intensifying systemic therapy for patients with high-risk localized disease has improved survival rates, whether the intensification is with an ablative approach or more intensive conventional chemotherapy.
However, high-dose therapy has become increasingly accepted for patients with metastatic disease or recurrence, particularly in Europe. TBI has been largely abandoned as part of the conditioning regimen. Many different regimens have been used, most a combination of melphalan with etoposide, busulfan, carboplatin, and thiotepa.87 The results of high-risk localized disease are usually lumped together with multifocal disease in the high-dose therapy reports, but the survival of patients with bone metastases at diagnosis appears much better than with standard therapy in some reports. EFS rates as high as 43% have been reported.88 The use of whole-body MRI to detect bone metastases followed by compartmental irradiation to doses up to 54 Gy and high-dose chemotherapy and stem cell rescue has also been reported to produce a 5-year survival rate of 45%.89 Although it was a small series, over half the patients had lung metastases and multiple bone metastases.
 TREATMENT AFTER RELAPSE
TREATMENT AFTER RELAPSE
The prognosis after relapse is poor, with a 5-year overall survival of only 13% for patients recurring after treatment in the CESS 81, CESS 86, and EICESS 92 studies. Patients experiencing either late (>2 years) or strictly localized relapses fared slightly better.1,90 Relapse occurring only at the local site warrants aggressive attempts at salvage.91 Patients who relapse only with lung metastases can sometimes be salvaged with additional chemotherapy and lung irradiation. Resecting the lung metastases, if there are fewer than four lesions, may also be beneficial.92 Patients with late pulmonary relapses fare better than those with early relapses.53 Patients who relapse with bone metastases, however, are essentially incurable with standard therapy.
 SEQUELAE OF TREATMENT
SEQUELAE OF TREATMENT
Ewing tumor has been reported to have an actuarial complication rate of 70% at 35 years.93 Paulino et al.35 reported that 10 (53%) of 19 patients receiving radiotherapy alone, four (25%) of 16 receiving surgery alone, and two (40%) of five undergoing combined surgery and radiation therapy had significant late effects.35 Neither study included loss of function related to planned surgical resection as a complication.
The most common skeletal complication of radiotherapy is abnormal growth and development of the irradiated tissues. Radiation can cause premature closure of active epiphyses, producing growth deficits and limb-length discrepancies. The degree of discrepancy depends on the radiation dose, patient’s age, and epiphysis radiated. Because 65% of leg growth is from the distal femoral (37%) and proximal tibial (28%) epiphyses, typical radiotherapy doses to the knee in boys younger than 14 years of age and girls younger than 12 years of age will usually cause a severe enough leg-length discrepancy to require intervention.94 Deficits of 2 to 6 cm can usually be managed by a shoe lift; larger deficits require surgical treatment.95
Approximately 15% of long-bone lesions develop pathologic fractures at some time in their course, 5% at diagnosis and 10% after radiation therapy, although approximately one-third of the latter are caused by tumor recurrence or occasionally a secondary malignancy.96 Whether the fracture is disease or treatment related, the most common site is the femur, particularly the proximal femur. Radiotherapy-related fractures usually occur within 24 months after treatment, but can be much later. Doses below 40 Gy using once-a-day doses of 1.8 to 2.0 Gy appear to have a very low risk, as does a hyperfractionated approach using 1.2 Gy twice a day from 50.4 to 55.2 Gy.48,96
Extremity weakness, decreased range of motion secondary to fibrosis, pain in the extremity (particularly in the early morning), discoloration of the skin, and lymphedema can also occur after radiotherapy, even with careful planning and sparing of an adequate strip of tissue.
The risk of secondary neoplasia at the site of the primary lesion is related to radiation therapy dose, with an increased risk at doses >60 Gy.5 Sarcomas, often osteosarcoma, are the most common second tumor, and the risk for megavoltage treatment has been reported as 1% to 4% at 20 years.44,97,98 With more intensive chemotherapy regimens and the increased use of etoposide, the risk of secondary leukemia is about 2%.54
 FUTURE CONSIDERATIONS
FUTURE CONSIDERATIONS
Although the 5-year survival rate of patients with ESFT appears to have improved since 1970, almost half of all patients diagnosed with Ewing tumor die of it within 10 years.69 Intensifying systemic treatment appears to improve survival as the recently completed (in 2005) COG trial evaluating the role of interval compression chemotherapy shows. High-dose chemotherapy and stem cell rescue may also produce better results, particularly for patients with metastatic disease at diagnosis, but the limits of intensification have probably been reached with the presently available drugs. Other drugs are being tested and newer approaches are also promising. The Ewing family of tumors is characterized by the t(11;22)(q24;q12) translocation that generates the EWS-FLI1 fusion transcription factor producing the malignant EWS cell. Because continued expression of EWS-FLI1 is believed to be critical for ESFT cell survival, clinically effective small-molecule inhibitors have been sought.
Mithramycin has been reported to inhibit expression of EWS-FLI1 downstream targets at the messenger RNA and protein levels.99 YK-4-279, a small molecule found by researchers at Georgetown University (Washington, DC) in a library of small molecules supplied by the National Cancer Institute, stops EWS-FLI1’s fusion protein from sticking to RNA helicase A, inducing apoptosis in ESFT cells, and reduces the growth of tumor cells.100 Other approaches are also being studied.101
These findings provide proof of principle that inhibiting the interaction of mutant cancer-specific transcription factors with the normal cellular binding partners required for their oncogenic activity provides a promising strategy for the development of uniquely effective, tumor-specific anticancer agents.
With regard to local therapy, the focus should be on improving function and decreasing the incidence of long-term sequelae. The use of image-guided intensity-modulated radiation therapy and proton therapy, as well as smaller margins, should spare significantly more normal tissues than older techniques, probably decreasing late effects. However, it is unlikely that refinements in local therapy with radiotherapy or surgery will significantly improve the cure rate for patients with apparent localized disease, although treating all metastases with radiation or surgery appears to be promising for high-risk patients.
 REFERENCES
REFERENCES
1. Cotterill SJ, Ahrens S, Paulussen M, et al. Prognostic factors in Ewing’s tumor of bone: analysis of 975 patients from the European Intergroup Cooperative Ewing’s Sarcoma Study Group. J Clin Oncol 2000;18:3108–3114.
2. Kissane JM, Askin FB, Foulkes M, et al. Ewing’s sarcoma of bone: clinicopathologic aspects of 303 cases from the Intergroup Ewing’s Sarcoma Study. Hum Pathol 1983;14:773–779.
3. Dunst J, Paulussen M, Jurgens H. Lung irradiation for Ewing’s sarcoma with pulmonary metastases at diagnosis: results of the CESS-studies. Strahlenther Onkol 1993;169:621–623.
4. Yock TI, Krailo M, Fryer CJ, et al. Local control in pelvic Ewing sarcoma: analysis from INT-0091–a report from the Children’s Oncology Group. J Clin Oncol 2006;24:3838–3843.
5. Ladenstein R, Hartmann O, Pinkerton R, et al. A multivariate and matched pair analysis on high-risk Ewing tumor (ET) patients treated by megatherapy (MGT) and stem cell reinfusion (SCR) in Europe (Meeting abstract). 1999 ASCO Annual Meeting, Vol 18. Atlanta, Georgia, 1999:555.
6. Perez CA, Tefft M, Nesbit ME Jr, et al. Radiation therapy in the multimodal management of Ewing’s sarcoma of bone: report of the Intergroup Ewing’s Sarcoma Study. Natl Cancer Inst Monogr 1981:263–271.
7. Gamberi G, Cocchi S, Benini S, et al. Molecular diagnosis in Ewing family tumors: the Rizzoli experience–222 consecutive cases in four years. J Mol Diagn 2011;13:313–324.
8. Hadfield MG, Quezado MM, Williams RL, et al. Ewing’s family of tumors involving structures related to the central nervous system: a review. Pediatr Dev Pathol 2000;3:203–210.
9. Sandberg AA, Bridge JA. Updates on cytogenetics and molecular genetics of bone and soft tissue tumors: Ewing sarcoma and peripheral primitive neuroectodermal tumors. Cancer Genet Cytogenet 2000;123:1–26.
10. Pollock BH, Krischer JP, Vietti TJ. Interval between symptom onset and diagnosis of pediatric solid tumors. J Pediatr 1991;119:725–732.
11. Ginsberg JP, de Alava E, Ladanyi M, et al. EWS-FLI1 and EWS-ERG gene fusions are associated with similar clinical phenotypes in Ewing’s sarcoma. J Clin Oncol 1999;17:1809–1814.
12. Edeiken J, Karasick D. Imaging in bone cancer. CA Cancer J Clin 1987;37:239–245.
13. Mentzel HJ, Kentouche K, Sauner D, et al. Comparison of whole-body STIR-MRI and 99mTc-methylene-diphosphonate scintigraphy in children with suspected multifocal bone lesions. Eur Radiol 2004;14:2297–2302.
14. Nakanishi K, Kobayashi M, Nakaguchi K, et al. Whole-body MRI for detecting metastatic bone tumor: diagnostic value of diffusion-weighted images. Magn Reson Med Sci 2007;6:147–155.
15. Daldrup-Link HE, Franzius C, Link TM, et al. Whole-body MR imaging for detection of bone metastases in children and young adults: comparison with skeletal scintigraphy and FDG PET. AJR Am J Roentgenol 2001;177:229–236.
16. Gyorke T, Zajic T, Lange A, et al. Impact of FDG PET for staging of Ewing sarcomas and primitive neuroectodermal tumours. Nucl Med Commun 2006;27:17–24.
17. Dobbs MD, Lowas SR, Hernanz-Schulman M, et al. Impact of abdominopelvic CT on Ewing sarcoma management. Acad Radiol 2010;17:1288–1291.
18. Pantou D, Tsarouha H, Papadopoulou A, et al. Cytogenetic profile of unknown primary tumors: clues for their pathogenesis and clinical management. Neoplasia 2003;5:23–31.
19. Ahrens S, Hoffmann C, Jabar S, et al. Evaluation of prognostic factors in a tumor volume-adapted treatment strategy for localized Ewing sarcoma of bone: the CESS 86 experience. Cooperative Ewing Sarcoma Study. Med Pediatr Oncol 1999;32:186–195.
20. Aparicio J, Munarriz B, Pastor M, et al. Long-term follow-up and prognostic factors in Ewing’s sarcoma. A multivariate analysis of 116 patients from a single institution. Oncology 1998;55:20–26.
21. Bacci G, Picci P, Mercuri M, et al. Predictive factors of histological response to primary chemotherapy in Ewing’s sarcoma. Acta Oncol 1998;37:671–676.
22. Barbieri E, Emiliani E, Zini G, et al. Combined therapy of localized Ewing’s sarcoma of bone: analysis of results in 100 patients. Int J Radiat Oncol Biol Phys 1990;19:1165–1170.
23. Fizazi K, Dohollou N, Blay JY, et al. Ewing’s family of tumors in adults: multivariate analysis of survival and long-term results of multimodality therapy in 182 patients. J Clin Oncol 1998;16:3736–3743.
24. Lee JA, Kim DH, Cho J, et al. Treatment outcome of Korean patients with localized Ewing sarcoma family of tumors: a single institution experience. Jpn J Clin Oncol 2011;41:776–782.
25. Marcus RB, Million RR. The effect of primary tumor size on the prognosis of Ewing’s sarcoma. Int J Radiat Oncol Biol Phys 1984;10(Suppl 2).
26. Sauer R, Jurgens H, Burgers JM, et al. Prognostic factors in the treatment of Ewing’s sarcoma. The Ewing’s Sarcoma Study Group of the German Society of Paediatric Oncology CESS 81. Radiother Oncol 1987;10:101–110.
27. Wunder JS, Paulian G, Huvos AG, et al. The histological response to chemotherapy as a predictor of the oncological outcome of operative treatment of Ewing sarcoma. J Bone Joint Surg Am 1998;80:1020–1033.
28. Hense HW, Ahrens S, Paulussen M, et al. Factors associated with tumor volume and primary metastases in Ewing tumors: results from the (EI)CESS studies. Ann Oncol 1999;10:1073–1077.
29. Dunst J, Ahrens S, Paulussen M, et al. Prognostic impact of tumor perfusion in MR-imaging studies in Ewing tumors. Strahlenther Onkol 2001;177:153–159.
30. Applebaum MA, Worch J, Matthay KK, et al. Clinical features and outcomes in patients with extraskeletal Ewing sarcoma. Cancer 2011;117:3027–3032.
31. Pradhan A, Grimer RJ, Spooner D, et al. Oncological outcomes of patients with Ewing’s sarcoma: is there a difference between skeletal and extra-skeletal Ewing’s sarcoma? J Bone Joint Surg Br 2011;93:531–536.
32. Marcus RB Jr, Berrey BH, Graham-Pole J, et al. The treatment of Ewing’s sarcoma of bone at the University of Florida: 1969 to 1998. Clin Orthop Relat Res 2002:290–297.
33. Gaston LL, Di Bella C, Slavin J, et al. 18F-FDG PET response to neoadjuvant chemotherapy for Ewing sarcoma and osteosarcoma are different. Skeletal Radiol 2011;40:1007–1015.
34. Fuchs B, Inwards CY, Janknecht R. Vascular endothelial growth factor expression is up-regulated by EWS-ETS oncoproteins and Sp1 and may represent an independent predictor of survival in Ewing’s sarcoma. Clin Cancer Res 2004;10:1344–1353.
35. Paulino AC, Nguyen TX, Mai WY. An analysis of primary site control and late effects according to local control modality in non-metastatic Ewing sarcoma. Pediatr Blood Cancer 2007;48:423–429.
36. Perbal B, Lazar N, Zambelli D, et al. Prognostic relevance of CCN3 in Ewing sarcoma. Hum Pathol 2009;40:1479–1486.
37. van Doorninck JA, Ji L, Schaub B, et al. Current treatment protocols have eliminated the prognostic advantage of type 1 fusions in Ewing sarcoma: a report from the Children’s Oncology Group. J Clin Oncol 2010;28:1989–1994.
38. Le Deley MC, Delattre O, Schaefer KL, et al. Impact of EWS-ETS fusion type on disease progression in Ewing’s sarcoma/peripheral primitive neuroectodermal tumor: prospective results from the cooperative Euro-E.W.I.N.G. 99 trial. J Clin Oncol 2010;28:1982–1988.
39. Avigad S, Cohen IJ, Zilberstein J, et al. The predictive potential of molecular detection in the nonmetastatic Ewing family of tumors. Cancer 2004;100:1053–1058.
40. Burgert EO Jr, Nesbit ME, Garnsey LA, et al. Multimodal therapy for the management of nonpelvic, localized Ewing’s sarcoma of bone: intergroup study IESS-II. J Clin Oncol 1990;8:1514–1524.
41. Evans RG, Nesbit ME, Gehan EA, et al. Multimodal therapy for the management of localized Ewing’s sarcoma of pelvic and sacral bones: a report from the second intergroup study. J Clin Oncol 1991;9:1173–1180.
42. Grier HE, Krailo MD, Tarbell NJ, et al. Addition of ifosfamide and etoposide to standard chemotherapy for Ewing’s sarcoma and primitive neuroectodermal tumor of bone. N Engl J Med 2003;348:694–701.
43. Horowitz ME, Kinsella TJ, Wexler LH, et al. Total-body irradiation and autologous bone marrow transplant in the treatment of high-risk Ewing’s sarcoma and rhabdomyosarcoma. J Clin Oncol 1993;11:1911–1918.
44. Nesbit ME Jr, Gehan EA, Burgert EO Jr, et al. Multimodal therapy for the management of primary, nonmetastatic Ewing’s sarcoma of bone: a long-term follow-up of the First Intergroup study. J Clin Oncol 1990;8:1664–1674.
45. Hayes FA, Thompson EI, Meyer WH, et al. Therapy for localized Ewing’s sarcoma of bone. J Clin Oncol 1989;7:208–213.
46. Oberlin O, Patte C, Demeocq F, et al. The response to initial chemotherapy as a prognostic factor in localized Ewing’s sarcoma. Eur J Cancer Clin Oncol 1985;21:463–467.
47. Scully SP, Temple HT, O’Keefe RJ, et al. Role of surgical resection in pelvic Ewing’s sarcoma. J Clin Oncol 1995;13:2336–2341.
48. Bolek TW, Marcus RB Jr, Mendenhall NP, et al. Local control and functional results after twice-daily radiotherapy for Ewing’s sarcoma of the extremities. Int J Radiat Oncol Biol Phys 1996;35:687–692.
49. Donaldson SS. Ewing sarcoma: radiation dose and target volume. Pediatr Blood Cancer 2004;42:471–476.
50. Korah MP, Esiashvili N, Mazewski CM, et al. Incidence, risks, and sequelae of posterior fossa syndrome in pediatric medulloblastoma. Int J Radiat Oncol Biol Phys 2010;77:106–112.
51. Marcus RB Jr, Cantor A, Heare TC, et al. Local control and function after twice-a-day radiotherapy for Ewing’s sarcoma of bone. Int J Radiat Oncol Biol Phys 1991;21:1509–1515.
52. Dunst J, Jurgens H, Sauer R, et al. Radiation therapy in Ewing’s sarcoma: an update of the CESS 86 trial. Int J Radiat Oncol Biol Phys 1995;32:919–930.
53. Craft A, Cotterill S, Malcolm A, et al. Ifosfamide-containing chemotherapy in Ewing’s sarcoma: the Second United Kingdom Children’s Cancer Study Group and the Medical Research Council Ewing’s Tumor Study. J Clin Oncol 1998;16:3628–3633.
54. Granowetter L. COG Public Report, 8/29/01. Available from: COG Statistical Center, 440 E Huntington Drive, Suite 300, Arcadia, CA 91006.
55. Schuck A, Ahrens S, Paulussen M, et al. Local therapy in localized Ewing tumors: results of 1058 patients treated in the CESS 81, CESS 86, and EICESS 92 trials. Int J Radiat Oncol Biol Phys 2003;55:168–177.
56. Rosito P, Mancini AF, Rondelli R, et al. Italian Cooperative Study for the treatment of children and young adults with localized Ewing sarcoma of bone: a preliminary report of 6 years of experience. Cancer 1999;86:421–428.
57. Krasin MJ, Rodriguez-Galindo C, Billups CA, et al. Definitive irradiation in multidisciplinary management of localized Ewing sarcoma family of tumors in pediatric patients: outcome and prognostic factors. Int J Radiat Oncol Biol Phys 2004;60:830–838.
58. Donaldson SS, Torrey M, Link MP, et al. A multidisciplinary study investigating radiotherapy in Ewing’s sarcoma: end results of POG #8346. Pediatric Oncology Group. Int J Radiat Oncol Biol Phys 1998;42:125–135.
59. Schuck A, Ahrens S, Konarzewska A, et al. Hemithorax irradiation for Ewing tumors of the chest wall. Int J Radiat Oncol Biol Phys 2002;54:830–838.
60. Shamberger RC, Grier HE, Weinstein HJ, et al. Chest wall tumors in infancy and childhood. Cancer 1989;63:774–785.
61. Schuck A, Ahrens S, von Schorlemer I, et al. Radiotherapy in Ewing tumors of the vertebrae: treatment results and local relapse analysis of the CESS 81/86 and EICESS 92 trials. Int J Radiat Oncol Biol Phys 2005;63:1562–1567.
62. Venkateswaran L, Rodriguez-Galindo C, Merchant TE, et al. Primary Ewing tumor of the vertebrae: clinical characteristics, prognostic factors, and outcome. Med Pediatr Oncol 2001;37:30–35.
63. Jereb B, Ong RL, Mohan M, et al. Redefined role of radiation in combined treatment of Ewing’s sarcoma. Pediatr Hematol Oncol 1986;3:111–118.
64. Ozaki T, Hillmann A, Hoffmann C, et al. Significance of surgical margin on the prognosis of patients with Ewing’s sarcoma. A report from the Cooperative Ewing’s Sarcoma Study. Cancer 1996;78:892–900.
65. Dunst J, Schuck A. Role of radiotherapy in Ewing tumors. Pediatr Blood Cancer 2004;42:465–470.
66. Merchant TE, Kushner BH, Sheldon JM, et al. Effect of low-dose radiation therapy when combined with surgical resection for Ewing sarcoma. Med Pediatr Oncol 1999;33:65–70.
67. Schuck A, Rube C, Konemann S, et al. Postoperative radiotherapy in the treatment of Ewing tumors: influence of the interval between surgery and radiotherapy. Strahlenther Onkol 2002;178:25–31.
68. Chan RC, Sutow WW, Lindberg RD, et al. Management and results of localized Ewing’s sarcoma. Cancer 1979;43:1001–1006.
69. Esiashvili N, Goodman M, Marcus RB. Incidence and survival of patients with Ewing sarcoma of bone over the past three decades based on surveillance epidemiology and end results data. 38th Annual Congress of International Society of Pediatric Oncology (SIOP), Geneva, Switzerland, 2006.
70. Gasparini M, Lombardi F, Ballerini E, et al. Long-term outcome of patients with monostotic Ewing’s sarcoma treated with combined modality. Med Pediatr Oncol 1994;23:406–412.
71. Paulussen M, Ahrens S, Dunst J, et al. Localized Ewing tumor of bone: final results of the cooperative Ewing’s Sarcoma Study CESS 86. J Clin Oncol 2001;19:1818–1829.
72. Womer RB, West DC, Krailo MD, et al. Randomized comparison of every-two-week v. every-three-week chemotherapy in Ewing sarcoma family tumors (ESFT). J Clin Oncol 2008;26:abstr 10504.
73. Paulussen M, Craft AW, Lewis I, et al. Results of the EICESS-92 Study: two randomized trials of Ewing’s sarcoma treatment–cyclophosphamide compared with ifosfamide in standard-risk patients and assessment of benefit of etoposide added to standard treatment in high-risk patients. J Clin Oncol 2008;26:4385–4393.
74. Raney RB, Asmar L, Newton WA Jr, et al. Ewing’s sarcoma of soft tissues in childhood: a report from the Intergroup Rhabdomyosarcoma Study, 1972 to 1991. J Clin Oncol 1997;15:574–582.
75. Miser JS, Krailo MD, Tarbell NJ, et al. Treatment of metastatic Ewing’s sarcoma or primitive neuroectodermal tumor of bone: evaluation of combination ifosfamide and etoposide–a Children’s Cancer Group and Pediatric Oncology Group study. J Clin Oncol 2004;22:2873–2876.
76. Paulussen M, Ahrens S, Craft AW, et al. Ewing’s tumors with primary lung metastases: survival analysis of 114 (European Intergroup) Cooperative Ewing’s Sarcoma Studies patients. J Clin Oncol 1998;16:3044–3052.
77. Spunt SL, McCarville MB, Kun LE, et al. Selective use of whole-lung irradiation for patients with Ewing sarcoma family tumors and pulmonary metastases at the time of diagnosis. J Pediatr Hematol Oncol 2001;23:93–98.
78. Haeusler J, Ranft A, Boelling T, et al. The value of local treatment in patients with primary, disseminated, multifocal Ewing sarcoma (PDMES). Cancer 2010;116:443–450.
79. La TH, Meyers PA, Wexler LH, et al. Radiation therapy for Ewing’s sarcoma: results from Memorial Sloan-Kettering in the modern era. Int J Radiat Oncol Biol Phys 2006;64:544–550.
80. Rodriguez-Galindo C, Navid F, Liu T, et al. Prognostic factors for local and distant control in Ewing sarcoma family of tumors. Ann Oncol 2008;19:814–820.
81. Paulino AC, Mai WY, Teh BS. Radiotherapy in metastatic Ewing sarcoma. Am J Clin Oncol 2012012 [Epub ahead of print].
82. Meyers PA, Krailo MD, Ladanyi M, et al. High-dose melphalan, etoposide, total-body irradiation, and autologous stem-cell reconstitution as consolidation therapy for high-risk Ewing’s sarcoma does not improve prognosis. J Clin Oncol 2001;19:2812–2820.
83. Kushner BH, Meyers PA. How effective is dose-intensive/myeloablative therapy against Ewing’s sarcoma/primitive neuroectodermal tumor metastatic to bone or bone marrow? The Memorial Sloan-Kettering experience and a literature review. J Clin Oncol 2001;19:870–880.
84. Pinkerton CR, Bataillard A, Guillo S, et al. Treatment strategies for metastatic Ewing’s sarcoma. Eur J Cancer 2001;37:1338–1344.
85. Burdach S, Meyer-Bahlburg A, Laws HJ, et al. High-dose therapy for patients with primary multifocal and early relapsed Ewing’s tumors: results of two consecutive regimens assessing the role of total-body irradiation. J Clin Oncol 2003;21:3072–3078.
86. Madero L, Munoz A, Sanchez de Toledo J, et al. Megatherapy in children with high-risk Ewing’s sarcoma in first complete remission. Bone Marrow Transplant 1998;21:795–799.
87. Rosenthal J, Pawlowska AB. High-dose chemotherapy and stem cell rescue for high-risk Ewing’s family of tumors. Expert Rev Anticancer Ther 2011;11:251–262.
88. Oberlin O, Rey A, Desfachelles AS, et al. Impact of high-dose busulfan plus melphalan as consolidation in metastatic Ewing tumors: a study by the Societe Francaise des Cancers de l’Enfant. J Clin Oncol 2006;24:3997–4002.
89. Burdach S, Thiel U, Schoniger M, et al. Total body MRI-governed involved compartment irradiation combined with high-dose chemotherapy and stem cell rescue improves long-term survival in Ewing tumor patients with multiple primary bone metastases. Bone Marrow Transplant 2010;45:483–489.
90. Stahl M, Ranft A, Paulussen M, et al. Risk of recurrence and survival after relapse in patients with Ewing sarcoma. Pediatr Blood Cancer 2011;57:549–553.
91. Hayes FA, Thompson EI, Kumar M, et al. Long-term survival in patients with Ewing’s sarcoma relapsing after completing therapy. Med Pediatr Oncol 1987;15:254–256.
92. Lanza LA, Miser JS, Pass HI, et al. The role of resection in the treatment of pulmonary metastases from Ewing’s sarcoma. J Thorac Cardiovasc Surg 1987;94:181–187.
93. Fuchs B, Valenzuela RG, Inwards C, et al. Complications in long-term survivors of Ewing sarcoma. Cancer 2003;98:2687–2692.
94. Anderson M, Green WT, Messner MB. Growth and predictions of growth in the lower extremities. J Bone Joint Surg Am 1963;45-A:1–14.
95. Moseley CF. Leg-length discrepancy. In: RT M, ed. Lovell and Winter’s pediatric orthopaedics, vol 2, 3rd ed. Philadelphia: Lippincott Williams & Wilkins, 1990:767–813.
96. Wagner LM, Neel MD, Pappo AS, et al. Fractures in pediatric Ewing sarcoma. J Pediatr Hematol Oncol 2001;23:568–571.
97. Kuttesch JF Jr, Wexler LH, Marcus RB, et al. Second malignancies after Ewing’s sarcoma: radiation dose-dependency of secondary sarcomas. J Clin Oncol 1996;14:2818–2825.
98. Tucker MA, D’Angio GJ, Boice JD Jr, et al. Bone sarcomas linked to radiotherapy and chemotherapy in children. N Engl J Med 1987;317:588–593.
99. Grohar PJ, Woldemichael GM, Griffin LB, et al. Identification of an inhibitor of the EWS-FLI1 oncogenic transcription factor by high-throughput screening. J Natl Cancer Inst 2011;103:962–978.
100. Erkizan HV, Kong Y, Merchant M, et al. A small molecule blocking oncogenic protein EWS-FLI1 interaction with RNA helicase A inhibits growth of Ewing’s sarcoma. Nat Med 2009;15:750–756.
101. Subbiah V, Anderson P. Targeted therapy of Ewing’s sarcoma. Sarcoma 2011;2011:686985.
< div class='tao-gold-member'>




