Introduction
The body must be protected from hypoglycemia because severe acute hypoglycemia can cause seizures, coma, and even death . On the other hand, chronic hyperglycemia is toxic to blood vessels, basement membranes, and proteins that become glycated . Therefore, the body must maintain the level of energy currency (glucose) in the bloodstream within narrow limits. This is a challenging task because the biochemical demands for glucose throughout the body can vary tremendously. Although there are four hormones specifically designed to raise glucose concentrations in the bloodstream [epinephrine, glucagon, cortisol, and growth hormone (GH)], there is only one hormone designed to specifically lower glucose concentrations: insulin. This chapter addresses the etiologies and diagnostic evaluations for hypoglycemia.
Clinical symptoms of hypoglycemia
In routine medical practice, hypoglycemia (low blood glucose) is commonly encountered in the immediate neonatal period [especially in premature infants and infants born to diabetic mothers (IDM)] and in insulin-treated diabetic patients who take excessive doses of insulin, skip meals, and/or exercise more than usual . Acute hypoglycemia produces adrenergic symptoms (e.g., tachycardia, palpitations, pounding heart, sweating, tremulousness, anxiety, nervousness, feeling cold, and sweating) and neuroglycopenia [low blood glucose affecting the brain: decreased consciousness, tiredness or drowsiness, faintness, confusion, blurred or double vision, hemiparesis, behavioral changes, dizziness, paresthesias (pins and needles feelings), incoordination, slurred speech, hunger, headache, seizures, coma, and even death].
Fasting hypoglycemia versus reactive hypoglycemia
Fasting (postabsorptive) hypoglycemia occurs most commonly in the morning following an overnight fast, between meals, or with exercise. Fasting hypoglycemia usually implies a serious medical condition that requires thorough evaluation.
Reactive hypoglycemia is also referred to as “postprandial” or “stimulative” hypoglycemia. This form of reputed hypoglycemia usually does not imply a serious disorder . The hypothesis behind “reactive” (or food-stimulated) hypoglycemia is that excessive release of insulin follows a large glucose load and causes hypoglycemia several hours after eating. Many patients with symptoms of reactive hypoglycemia regularly miss meals and/or do not eat a balanced diet (e.g., ~55%–60% carbohydrate, ~15% protein, ≤30% fat). The diagnosis of reactive hypoglycemia is sometimes inappropriately pursued by performing a 5-h oral glucose tolerance test (OGTT). When patients with hypoglycemic-like symptoms manifest a glucose of <40 mg/dL in the later stages of the OGTT, reactive hypoglycemia is diagnosed. However, ≥10% of adults display glucose values of <50 mg/dL at 3 h or more after an oral glucose challenge. In such general population studies, there has been no correlation between the adrenergic symptoms of reactive hypoglycemia and glucose values <50 mg/dL 3–5 h after the oral glucose load. For this reason, most endocrinologists do not consider reactive hypoglycemia to be a disease. Clinicians should therefore be discouraged from performing 5-h OGTTs. Successful treatment of the symptoms of reactive hypoglycemia involves achieving healthy eating patterns and regular meals.
The only hypoglycemic disorder that is truly “reactive” involves the “dumping” syndrome, where food (often hyperosmolar) rapidly transits from the stomach into the small intestine and large amounts of glucose are rapidly absorbed . Subsequently, there is a surge of glucose-driven and incretin-driven insulin release that does, in fact, lead to true “reactive” hypoglycemia . This form of “reactive” hypoglycemia can be treated with smaller, more frequent meals. Hyperinsulinism following gastric bypass surgery is another cause of reactive hypoglycemia .
Diagnosis of hypoglycemia
The diagnosis of hypoglycemia is established when three criteria (“Whipple’s triad”) are documented: (1) symptoms compatible with hypoglycemia are substantiated; (2) there is laboratory confirmation of a low blood glucose concentration at the time of symptoms (serum or plasma glucose <45 mg/dL); and (3) the symptoms are relieved promptly with the rapid administration of glucose . Because self-blood glucose monitoring systems are usually not accurate at values <40–60 mg/dL, the glucose determination must be performed on a sample obtained by venipuncture using instrumentation with an accurate and precise lower limit of detection, ideally, of 10 mg/dL or less.
Biochemical definition of hypoglycemia
Hypoglycemia is biochemically defined as a venous whole-blood glucose of <40 mg/dL or a venous plasma or serum glucose of <45 mg/dL (note: whole-blood glucose is ~15% lower than plasma or serum glucose because of the nonaqueous constituents of whole blood). Some references set a cutoff for hypoglycemia as a serum or plasma glucose <40 mg/dL. Other experts judge any serum or plasma glucose concentration of <50 mg/dL as representing hypoglycemia.
Because glucose concentrations in plasma or serum are not affected by changes in the hematocrit, plasma or serum is the preferred specimen for glucose analysis. Although newborns, especially premature infants, commonly display glucose values of 20–40 mg/dL or less, these values are considered to be potentially harmful . Therefore, the definition of hypoglycemia described above should apply to individuals of all ages.
Pseudohypoglycemia (artifactual hypoglycemia) occurs most often when the measured blood glucose is artifactually depressed because of delayed analysis . Because cellular elements of the bloodstream (e.g., white blood cells and red blood cells) consume glucose, unless samples are to be assayed within 1 h of phlebotomy, blood should be drawn into gray-top tubes containing NaF to inhibit glycolysis (which takes ~1 h for complete inhibition). Potassium chromate serves as an anticoagulant in gray-top tubes. Without the addition of NaF, serum or plasma glucose will continue to fall by ~2%–3% h after the first-hour postphlebotomy. Samples placed in NaF tubes will be stable at room temperature for 24 h. High white blood cell counts (e.g., chronic myelocytic leukemia) can produce more rapid rates of decline in blood glucose . Artifactual hypoglycemia has also been described in persons with polycythemia vera . Factitious hypoglycemia is self-induced hypoglycemia (e.g., a person with diabetes intentionally takes an excessive dose of insulin with the intention of producing hypoglycemia).
Causes of hypoglycemia
In the absence of a recognized cause of hypoglycemia (e.g., an IDM or a diabetic patient who took a known overdose of insulin), the initial challenge is to determine whether the patient has hyperinsulinemic hypoglycemia or nonhyperinsulinemic hypoglycemia. Assuming that hypoglycemia has been biochemically confirmed, the age of the patient has a major influence on the likely causes of hypoglycemia ( Table 7.1 ). Therefore, the age of the patient should influence the laboratory tests that are ordered and performed. For example, in individuals who first develop hypoglycemia after the ages of 4 or 5 years, studies in search of inborn errors of metabolism should not generally be pursued.
| Neonate | Child | Adult | |
|---|---|---|---|
| Hyperinsulinemia* | ++ | ++ | +++ |
| Drugs | + | +++ | +++ |
| Hormone deficiency | ++ | ++ | + |
| Inborn errors | ++ | + | − |
| Liver disease | ++ | ++ | ++ |
| Renal disease | − | − | − |
| Extrapancreatic neoplasm | − | − | + |
| Critical or severe illness | +++ | ++ | + |
| Neonatal hypoglycemia | +++ | N/A | N/A |
| Ketotic hypoglycemia | N/A | ++ | N/A |
| Severe malnutrition | + | + | + |
Diagnostic workup for hypoglycemia
Evaluation of acute hypoglycemia
Should the patient suffer an acute episode of hypoglycemia (e.g., the patient arrives in the emergency room in coma or postictal and is found to be hypoglycemic), Table 7.2 lists the studies that should be considered. If possible, more than one blood sample should be obtained at this time [e.g., the initial sample and a sample drawn just before intravenous (IV) glucose or glucagon are administered] . Tests for the evaluation of hypoglycemia can be divided into routine versus optional, and those tests that should be obtained at the time of acute hypoglycemia versus those tests that need not be obtained at the time of acute hypoglycemia. The optional tests are dependent upon the patient’s age, history, physical examination, and likely causes of hypoglycemia depending upon the specific clinical situation. For example, if in the initial evaluation, the patient is found to have liver failure, other causes of hypoglycemia are less likely and need not immediately be pursued. These guidelines for obtaining samples also apply to prolonged fasts.
| Test | Indication |
|---|---|
| Routine measurements in adults and children at the time of acute hypoglycemia | |
| Plasma glucose | Measured to diagnose hypoglycemia; decreased value is necessary for the diagnosis of hypoglycemia (gray-top tube is the preferred sample) |
| βHB (and/or) urine ketones | Assessment of ketosis; hyperinsulinism can suppress ketosis |
| Insulin | Measured to rule out hyperinsulinism (consider proinsulin and C-peptide measurements) |
| Routine measurements in infants and young children at the time of acute hypoglycemia | |
| Lactic acid | Assessment for lactic acidosis; measurement in adults is optional (unless otherwise measured, as in sepsis) |
| Urine-reducing substances | Measured to exclude certain inborn errors of carbohydrate metabolism; measure in all infants (e.g., ≤2 years): measure at a time when the infant is on their normal diet |
| Optional measurements at the time of acute hypoglycemia | |
| Drug screen/ethanol | Performed to rule out intoxication; perform blood/urine screening when intoxication cannot be otherwise excluded; screen for sulfonylureas and meglitinides when indicated |
| Cortisol a | Measured to rule out hypocortisolism; hold sample for cortisol measurement if indicated |
| Growth hormone (GH) | Measured to rule out GH deficiency; hold sample for GH measurement if indicated |
| C-peptide | Measured to confirm that hyperinsulinism is endogenous; C-peptide is elevated in insulinoma and sulfonylurea/meglitinide toxicity, with exogenous insulin administration, C-peptide is suppressed |
| Branched-chain amino acids | Measured to document the biochemical effects of hyperinsulinism; rarely needed; helpful in confirming “functional” hyperinsulinism when insulin levels are low but other agents cause hypoglycemia (e.g., IGF-II excess or agonistic antiinsulin receptor autoantibodies) |
| Free fatty acids (FFAs) | Measured to document the biochemical effects of hyperinsulinism or suspected fatty acid oxidation disorders (FAOD) |
| Urine dicarboxylic acids | Measured when an aminoacidopathy or FAOD are suspected |
| Arterial blood gases b | Measured to assess acid/base balance; acidosis is common in many disorders that cause hypoglycemia |
| <SPAN role=presentation tabIndex=0 id=MathJax-Element-1-Frame class=MathJax style="POSITION: relative" data-mathml='HCO3−’>HCO−3HCO3− HCO 3 − , Cl − , urine pH | Measured to assess acid/base balance; acidosis is common in many disorders that cause hypoglycemia |
| Routine measurements not required at the time of acute hypoglycemia | |
| Liver function c | Measured to rule out liver disease |
| Renal function d | Measured to rule out kidney disease |
| Optional measurements not required at the time of acute hypoglycemia | |
| Thyroid studies e | Measured to rule out thyroid dysfunction; measure when thyroid disease is clinically suspected |
| Serum/plasma amino acids | Measured to rule out an aminoacidopathy |
| Uric acid | Measured when inborn errors of metabolism are a consideration; often elevated when lactate is elevated |
| Cholesterol/triglycerides | Measured when inborn errors of metabolism are a consideration; elevated in certain glycogen storage diseases |
| Creatine kinase (CK) | Measured when myopathy is suspected; elevated in certain glycogen storage diseases |
| Carnitine | Measured when FAOD is suspected; low in cases of carnitine deficiency |
c ALT, alkaline phosphatase, total bilirubin, direct bilirubin, albumin, total protein, consider prothrombin time (PT), and AST.
d Creatinine, estimated glomerular filtration rate, blood urea nitrogen (BUN), urinalysis.
In the absence of an acute hypoglycemic episode, the best time to test for hypoglycemia is in the morning after an overnight fast. Plasma glucose values of <45 mg/dL are consistent with hypoglycemia, whereas plasma glucose concentrations of 45–60 mg/dL are suggestive of hypoglycemia. In either case, in persons presenting with symptoms suggestive of hypoglycemia, further evaluation when the glucose is ≤60 mg/dL is indicated (e.g., a prolonged fast should be undertaken). When hypoglycemia is a serious clinical consideration, several fasting blood glucose determinations are warranted before hypoglycemia is excluded. Plasma glucose concentrations >60 mg/dL are normal, when present on several occasions (in the absence of any values ≤60 mg/dL) rule against fasting hypoglycemia.
This discussion of plasma glucose cut points exposes areas that demand clarification. The typical “normal range” for fasting plasma glucose is 70–99 mg/dL. Yet, we have defined a plasma glucose of >60 mg/dL as being “normal” and a plasma glucose of <45 mg/dL as being pathologically low. Therefore, we need a better understanding of, and a term for, this borderline range of plasma glucose values that are of from 45 to 60 or 69 mg/dL [analogous to the description of “prediabetes” (see “Evaluation of hyperglycemia” in Chapter: 8)].
Prolonged fasts in the evaluation of hypoglycemia
A prolonged fast lasting up to 72 h in adults, or at least 24–36 h in children, may be required to determine whether or not hypoglycemia is actually present. In adults with proven insulinoma, fasting for 48 h is usually sufficient to elicit hypoglycemia . In children with ketotic hypoglycemia, fasting for as little as 8 h may precipitate hypoglycemia . “Ketotic hypoglycemia” likely reflects the limited biochemical capacities of young children to maintain normoglycemia because of their proportionately smaller muscle mass (as a source of gluconeogenic precursors) and higher ratio of brain size to liver size [e.g., the ratio of the glucose consumer (the brain) to the glucose producer (the liver) compared to adults]. Normal children do not become hypoglycemic with short-term fasting .
Prolonged fasts are performed in the hospital, so that purported symptoms and signs of hypoglycemia can be carefully recorded. As well, plasma glucose concentrations can be regularly measured, blood samples can be obtained for analysis, IV glucose is readily available for emergency administration, and IV glucagon can be administered at the termination of the fast to assess the degree of glucose reserve [e.g., the amount of glycogen present in the liver that can be liberated by glucagon injection at the end of the fast (see below)]. During the planned fast, the subject is given nothing by mouth except for free access to water or a glucose-free electrolyte solution.
At the beginning of the prolonged fast (before retiring for sleep), an IV line is introduced into a vein of sufficient size, so that blood can be periodically withdrawn for analysis. The line is flushed with a heparinized saline solution (e.g., 100 units/mL) and clamped. Prior to drawing subsequent samples, 3–5 mL of blood is withdrawn to clear the line of the flush solution (e.g., drawing a “blank”). Unless the patient is anemic, the “blank” blood sample can be discarded.
Blood must be drawn at the beginning of the fast, whose analysis provides a baseline plasma glucose value. Because adrenergic symptoms of hypoglycemia can become attenuated in some individuals who suffer chronic and recurrent hypoglycemia, during the fast, the plasma glucose must be measured regularly (e.g., every 4 h) by the central laboratory. Between these 4-h measurements, finger stick capillary glucose can be measured with a meter to monitor the course of the fast. If any of the capillary glucose values show a significant decline (e.g., decrease of 10 mg/dL or more), a confirmatory venous plasma glucose should be drawn from the indwelling catheter. Recall that capillary glucose values require correction to serum levels (capillary glucose×1.15 ~=serum glucose) because capillary and whole-blood glucose values are ~15% lower than venous plasma glucose values. Alternatively, if the point-of-care testing device reports the capillary glucose levels as plasma values, no adjustment is required.
During the fast, all urine samples are tested for ketones. Ketonuria is a normal response to fasting over prolonged periods of time (e.g., ≥18–24 h of fasting). Once the plasma glucose drops are below 70 mg/dL, venous draws can be increased to every 2 h. The fast is continued until frank symptoms of hypoglycemia are present or the venous plasma glucose is persistently <45 mg/dL (e.g., for at least 30 min). If this does not occur by the conclusion of the fast, fasting hypoglycemia is excluded, and further studies are not indicated.
During the fast, once symptoms of hypoglycemia initially occur or the venous plasma glucose initially falls below 45 mg/dL, blood should be drawn for clinical studies ( Table 7.2 ). This is a critical time for sampling when the laboratory results are most informative. Blood should be drawn again ~30 min later and the plasma glucose measurement is repeated. Because the inclusion or exclusion of hyperinsulinism is key to the differential diagnosis of hypoglycemia, insulin should essentially always be measured at these critical times (see below).
At the termination of the fast, if biochemical hypoglycemia is documented, IV glucagon (1 mg or 0.03 mg/kg, minimum dose in children is usually 0.5 mg) is administered. Blood for glucose measurement is drawn approximately every 10–15 min thereafter for up to 45 min, and blood is again stored for future analyses as described above. Interpretation of the plasma glucose postglucagon is discussed below. Obviously, the tests actually run depend upon the age of the patient, clinical history, physical examination, and clinical suspicion. The laboratory should be able to process and hold specimens until the physician decides which studies are required. Physicians should be encouraged to consult their clinical pathologists or clinical chemists about such matters.
Testing strategy for evaluation of hypoglycemia
In the differential diagnosis of hypoglycemia, the first diagnostic assessment concerns whether hyperinsulinism is present or absent at the time of hypoglycemia. This divides the etiologies of hypoglycemia into hyperinsulinemic hypoglycemia ( Fig. 7.1 ) and nonhyperinsulinemic hypoglycemia ( Fig. 7.2 ).
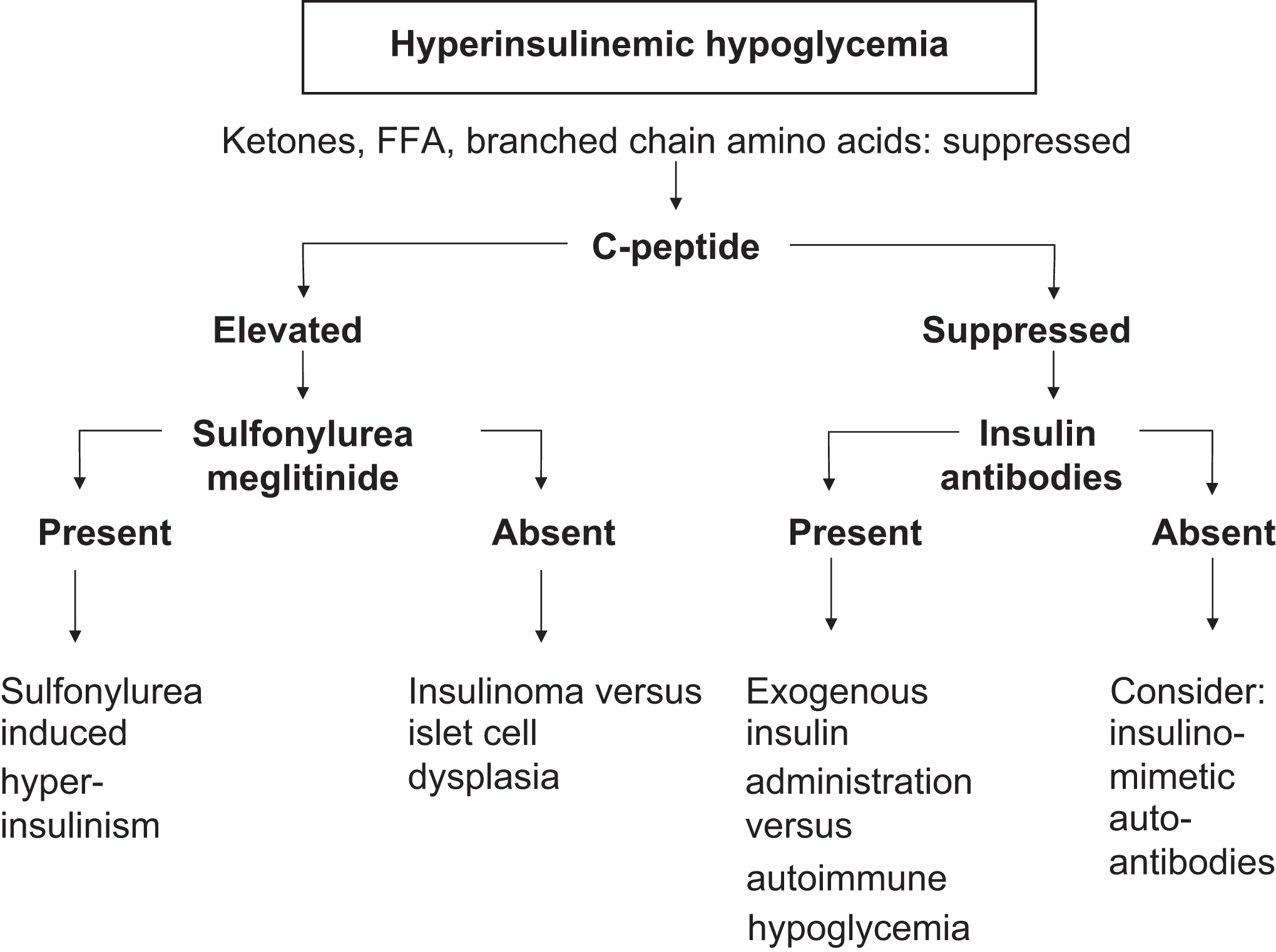
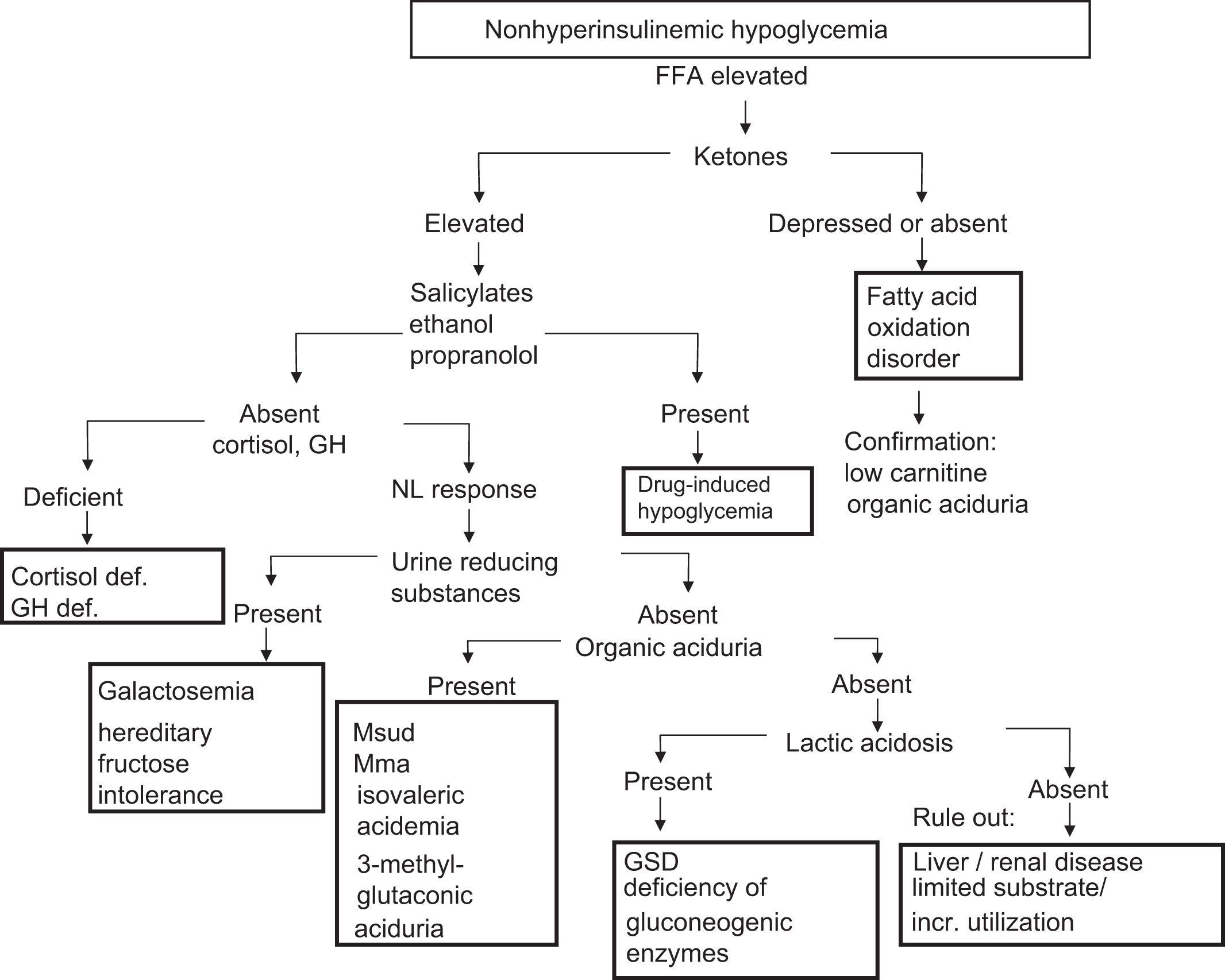
Hypoglycemia syndromes with hyperinsulinism
To determine whether hyperinsulinemic hypoglycemia is present, the clinician must assess two aspects of the patient’s presentation: (1) the insulin concentration relative to the glucose concentration and (2) whether other biochemical findings [e.g., urine ketones, β-hydroxybutyrate (βHB), free fatty acids (FFAs), branched-chain amino acids] are consistent with hyperinsulinism. Even when the insulin concentration is below the lower limit of the reference interval for an individual in the fasting state, the absolute insulin concentration may still be inappropriately elevated for the patient’s plasma glucose concentration. Some references suggest that measurements of C-peptide or proinsulin may provide better information than insulin measurements in determining whether hyperinsulinism is present .
“Functional” hyperinsulinism (e.g., increased insulin action without an inappropriate elevation in the insulin concentration) can also occur when there is an insulin surrogate such as an elevated insulin-like growth factor (IGF) II concentration from a tumor or an agonistic antiinsulin receptor autoantibody (insulinomimetic autoantibody) .
Some clinicians find that calculating the insulin/glucose ratio is helpful in diagnosing hyperinsulinism. If the insulin/glucose ratio is >0.25–0.30 (units: 100 uIU/mg), pathologic hyperinsulinism is diagnosed (to convert uIU/mL to pmol/L, multiply uIU/mL by 6.0). Other endocrinologists state that any measurable insulin in the setting of hypoglycemia is inappropriately elevated. However, this may not be appropriate as the lower limit of detection for insulin immunoassays is declining with advances in electrochemiluminescent technologies.
The laboratorian and clinician must also understand the specificity of the lab’s insulin assay. There are many recombinant DNA-derived insulins with altered sequences and compositions that are available for patient use. Not all of these insulins react on a 1 to 1 molar basis relative to human insulin in various insulin immunoassays. Certain insulin immunoassays only detect human insulin. Even if this is the case, the assay does not discriminate endogenous human insulin from injected human insulin. To further complicate the situation is the finding that insulin antibodies (IA) develop in more than 90% of insulin-treated patients regardless of the nature of the injected insulin . These IA act as circulating binding proteins and raise the total insulin concentration. However, since most of this insulin is bound in the plasma to the IA, only the “free” (unbound) insulin is bioactive. Also problematic is that there is no reliable way to measure free insulin (those assays are not robust and are poorly documented in the literature) . In the setting of IA, because plasma insulin measurements do not reflect the level of bioactive insulin, insulin should not be measured in insulin-treated patients. IA can appear within 2 wk of beginning insulin treatment.
An elevated proinsulin level or proinsulin/total insulin ratio in the face of hypoglycemia can also suggest hyperinsulinism. However, this requires an immunoassay selective for proinsulin. An amended insulin/glucose ratio was proposed in the 1970s: ([insulin]×100)/(glucose—30 mg/dL) (abnormal, >50). However, some authors believe that the best assessment for hyperinsulinism is the measurement of absolute insulin concentration during hypoglycemia (as noted above). Current generation automated electrochemiluminescent (ECL) insulin assays have very low to lower limits of detection for insulin.
With hyperinsulinism, the following laboratory findings are common because of the suppression of lipolysis, ketogenesis, and proteolysis by hyperinsulinism:
- •
urine negative for ketones;
- •
normal βHB (βHB is not elevated);
- •
normal FFA concentrations (FFAs are not elevated); and
- •
normal branched-chain amino acids (branched-chain amino acids are not elevated).
These findings reflect the suppressive effect of hyperinsulinism on the generation of alternative fuels (e.g., ketones) and the suppressive effect of hyperinsulinism on the liberation of amino acids from muscle (e.g., branched-chain amino acids) as substrate for gluconeogenesis . FFA and branched-chain amino acid determinations will usually be available only at reference laboratories. The absence of urine ketones and normal βHB in the face of hypoglycemia should alert the clinician and the laboratorian to the possibility of hyperinsulinism. However, by itself, the presence of urine ketones does not exclude hyperinsulinism. Lactic acidosis or elevated lactate does not occur as part of the hyperinsulinism syndromes. As will be discussed, lactic acidosis is seen with certain inborn errors of metabolism involving, for example, defective gluconeogenesis or glycogenolysis (e.g., glycogen storage disease [GSD]).
Another clinical aid in making the diagnosis of hyperinsulinism is the plasma glucose response to glucagon administration. This is usually done at the completion of a period of fasting when hypoglycemia has been confirmed or with acute hypoglycemia if glucagon is available for emergency administration (and the glucagon also serves as potential treatment for hypoglycemia). When hyperinsulinism is present, exogenous glucagon administered in a pharmacologic dose (e.g., 1 mg intramuscularly or IV) reverses hyperinsulinemic inhibition of glycogenolysis (as well as gluconeogenesis) and promotes rapid glycogenolysis, raising the plasma glucose of ≥30–40 mg/dL within ~45 min. With nonhyperinsulinemic hypoglycemia, the plasma glucose response to glucagon is <30–40 mg/dL and can be delayed or absent.
Hyperinsulinism with elevated C-peptide
If hyperinsulinism is diagnosed, the source of the insulin must be identified. Was the insulin released from the β cells of the islets of Langerhans, or was the insulin injected? When the pancreatic β cell is the source of hyperinsulinism (e.g., elevated C-peptide and insulin), the β cell may be functioning autonomously (e.g., insulinoma) or the β cell may be stimulated to oversecrete insulin by drugs , or, rarely, by β cell agonist autoantibodies . A recent paper reported the abuse of sulfonylureas to induce a euphoric “hypoglycemic rush” .
The differential diagnosis of autonomous β cell hyperfunction in neonates includes transient hyperinsulinism in small for gestational age (SGA) infants and transient hyperinsulinism secondary to exposure to maternal hyperglycemia (e.g., IDM) or erythroblastosis fetalis . Hypoglycemia in newborns limited to the first day of life usually represents “immaturity of fasting adaptation” due to a transient delay in the liver’s ability to carry out gluconeogenesis and ketogenesis .
Guidelines on the management of neonatal hypoglycemia have been published by the American Academy of Pediatrics and the Pediatric Endocrine Society. Hypoglycemia that persists over the first few days of life can result from maternal factors such as maternal diabetes, IV glucose administration to the mother during labor (that elicits hyperinsulinism), terbutaline (a β2 agonist) use, or propranolol (a beta-blocker) use . Hypoglycemia that lasts more than 2 d (but is not permanent and usually resolves by the age of 2–3 months) can be due to perinatal stress that causes transient hyperinsulinism (e.g., birth asphyxia , toxemia, erythroblastosis fetalis , SGA infant, or premature infant), hypopituitarism, or the Beckwith–Wiedemann syndrome.
Neonatal hyperinsulinism
When hypoglycemia is persistent and hyperinsulinism is confirmed, several inborn errors must be considered, including loss-of-function mutations where the β cell potassium channel (K ATP channel) is closed inducing persistent β cell depolarization and insulin release and gain-of-function mutations in glutamine dehydrogenase (GDH) or glucokinase .
K ATP channel mutations can be inherited as autosomal dominant or recessive traits. The K ATP channel is composed of four potassium channel subunits (Kir6.2) surrounded by four regulatory subunits [sulfonylurea receptor (SUR) 1]. Kir6.2 is encoded by the gene KCNJ11 (potassium inwardly rectifying channel, subfamily J, number 11). SUR1 is encoded by the gene ABCC8 (ATP binding cassette subfamily C member 8). K ATP channel mutations can cause diffuse β cell hyperplasia (requiring near-total pancreatectomy) or focal β cell hyperplasia (that can be managed through resection of a focal lesion) . GDH is encoded by GLUD1 . Many new and rare causes of hyperinsulinemic hypoglycemia in infants have been described in the last 10 years ( Table 7.3 ) .
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


