32.1
Introduction
The skeleton is a highly unusual organ because of the large number of hormones required to regulate its growth, maturation, and maintenance. Among the most important are the sex steroids—estrogen (E) and testosterone (T). In pioneering studies in the 1940s, Fuller Albright demonstrated that postmenopausal osteoporotic women are in negative calcium balance and that this could be corrected by E replacement. Although Albright considered osteoporosis in aging women to be due to impaired bone formation induced by menopause, more specific studies using bone biopsy analysis or radiocalcium kinetics subsequently demonstrated that postmenopausal osteoporosis results from an increase in bone resorption that is not compensated for by a concurrent increase in bone formation. With the advent of bone densitometry in the 1970s and 1980s, it was demonstrated that age-related bone loss in postmenopausal women could be normalized by E replacement, thus fulfilling Koch’s postulates. The major competing theory for the causation of osteoporosis during this interval was the effect of age-related decreases in calcium absorption and increases in renal calcium losses. With the unusual exception of clinically evident hypogonadism, osteoporosis in most aging men was thought to be caused by factors unrelated to sex steroids (see Riggs et al. for review of the historical background).
In this chapter, we review the actions of E on skeletal and extraskeletal calcium metabolism and show how the loss of these actions accounts for a significant proportion of age-related osteoporosis in both sexes. T plays a complementary but, at least in women, largely subsidiary role to E in regulating bone and calcium metabolism. We also touch briefly on major processes that contribute to the risk for osteoporosis but are not clearly attributable to E deficiency, including the finding of substantial trabecular bone loss in young adult women and men, as well as the role of aging mechanisms independent of E.
32.2
Age- and sex-specific skeletal changes
32.2.1
Measurement methods
Because of wide availability, lower costs, and lower radiation exposure, in almost all clinical studies and in most epidemiologic studies, bone measurements have been made using dual energy X-ray absorptiometry (DXA). However, DXA measures areal bone mineral density (aBMD) that substantially overestimates true volumetric BMD (vBMD). In contrast, quantitative computerized tomography (QCT) measures vBMD directly and also is capable of measuring bone size, shape, and the relative proportion of cortical and trabecular bone, key determinants of bone strength. Moreover, high resolution peripheral QCT (HRpQCT) methods have been developed that are capable of assessing trabecular bone structure noninvasively.
32.2.2
Patterns of change with growth and maturation
Skeletal size and vBMD are similar in prepubertal girls and boys. Between the onset of puberty and young adulthood, however, bone size triples and areal bone density doubles in boys, but in girls bone size only doubles with similar increases in areal bone density as boys . The rates of increase in statural height and bone remodeling are greatest in early puberty and then decline progressively until epiphyseal closure . In contrast, maximal increases in vBMD occur 2 years later—at menarche in girls and in late puberty in boys. The pattern of growth of boys differs from that of girls in two ways: boys have 2 more years of prepubertal growth because of their later puberty (age 14 years, rather than age 12 years as in girls), and their pubertal growth spurt lasts for 4 years rather than the 3 years that it lasts in girls. These differences largely account for the 10% greater statural height and the 30% greater peak bone mass achieved by males. For the most part the greater bone mass in males is due to their greater bone size rather than to differences in vBMD.
The increase in bone mass during pubertal growth occurs mainly by two processes that increase the length and width of bones, especially long bones. Linear bone growth occurs by ossification of the endochondral growth plates and continues until the growth plates fuse. Radial bone growth occurs by increased periosteal apposition, which is greater in males than in females. Periosteal apposition continues throughout life, but at a much slower rate after completion of puberty. During pubertal growth, periosteal apposition occurs mainly by modeling (bone formation on the periosteal surface that is not preceded by bone resorption). In pubertal males, there is a net increase in endosteal resorption that increases the marrow cavity size, whereas in pubertal females, endosteal resorption is decreased. These periosteal and endosteal changes account for the sexual dimorphism of the adult skeleton, especially in the long bones . Puberty is terminated by epiphyseal plate closure, by which time bone mass has reached about 90%–95% of eventual peak. A process termed “consolidation” then brings the skeleton to its maximal values by continued rapid periosteal apposition and, possibly, also by trabecular thickening. How long consolidation continues is disputed: it probably is completed within a few years although some reports suggest that it may continue into the third decade. The importance of bone mass acquisition during puberty is highlighted by the fact that the disorders of puberty (e.g., delayed puberty) are associated with potentially lifelong bone deficits.
Kirmani et al. have used HRpQCT to define trabecular and structural changes at the distal radius during growth in girls and boys spanning the age range of 6–21 years. They found that, at least at this site, trabecular parameters (bone volume fraction, trabecular number, and thickness) did not change in girls but increased in boys from late puberty onward. Cortical thickness and density decreased from pre- to midpuberty in girls but were unchanged in boys, before rising to higher levels at the end of puberty in both sexes. Total bone strength, assessed using micro-finite element models, increased with age in both sexes, with boys having greater bone strength than girls at the end of puberty. Interestingly, the proportion of load borne by cortical bone, and the ratio of cortical to trabecular bone volume, decreased transiently during mid- to late-puberty in both sexes, with apparent cortical porosity peaking during this time. These changes mirrored the incidence of distal forearm fractures in prior studies and suggested that regional deficits in cortical bone may underlie the adolescent peak in forearm fractures. In subsequent studies, Farr et al. demonstrated that compared with sex-matched controls, boys and girls with a mild-trauma distal forearm fracture (e.g., fall from standing height) showed significant deficits at the distal radius in failure load (−13% and −11%, respectively; P <.05) and had higher (worse) fall load-to-strength ratios (both +10%; P <.05 for boys and P =.06 for girls). In addition, boys and girls with a mild-trauma distal forearm fracture had significant reductions in cortical area (−26% and −23%, respectively; P <.01) and cortical thickness (−14% and −13%, respectively; P <.01) compared with controls.
32.2.3
Patterns of change with age in women
32.2.3.1
Overview
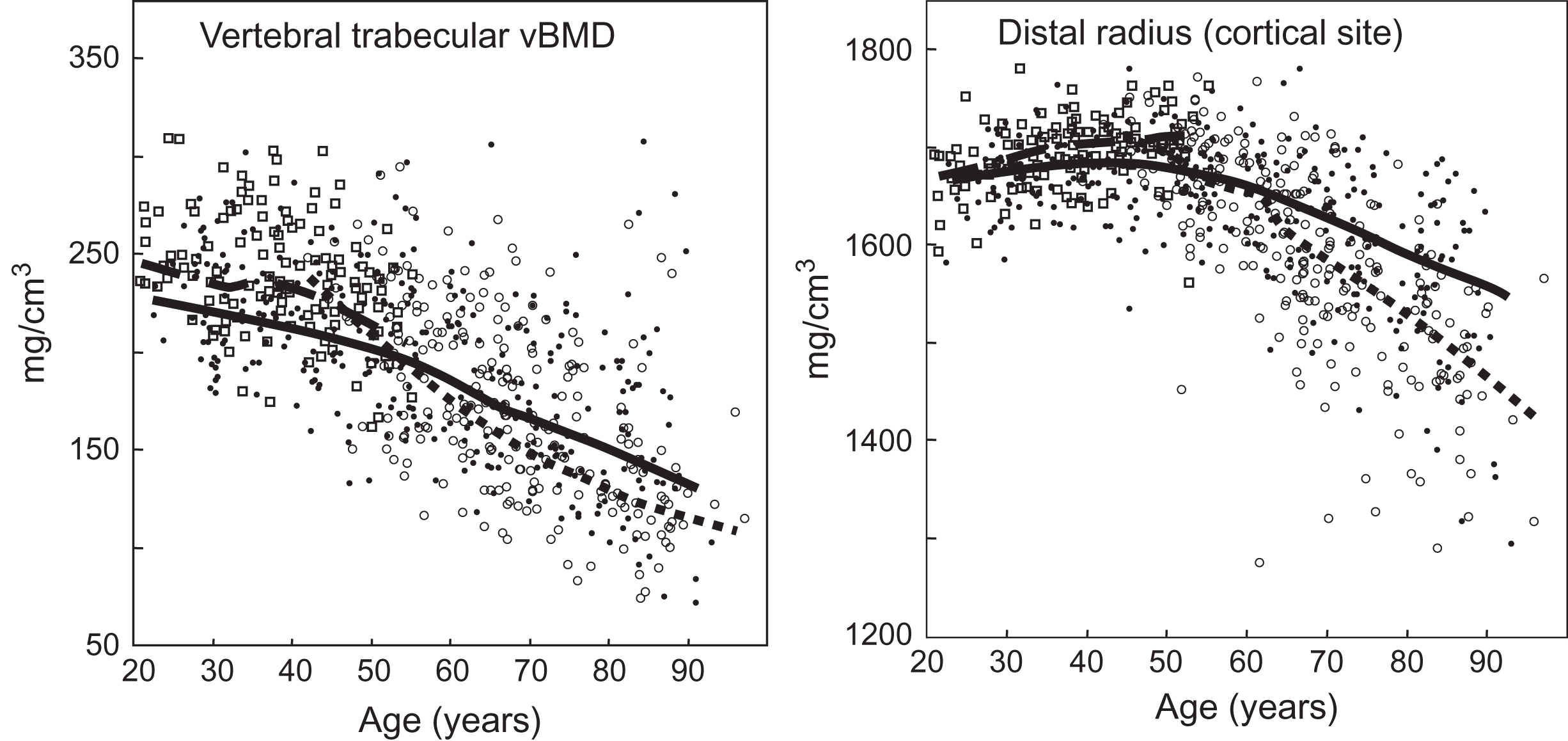
When vBMD or rates of change in vBMD as assessed by QCT are plotted over life, three phases of bone loss can be demonstrated in women: (1) an early phase of trabecular bone loss in late premenopausal women, (2) a transient phase beginning at menopause of rapid cortical bone loss and an acceleration of preexisting trabecular bone loss, and (3) a subsequent slower phase of loss involving similar amounts of cortical and trabecular bone that continues indefinitely ( Fig. 32.1 ). In addition to the overall bone loss that decreases bone strength, there are age-related changes that tend to sustain bone strength, thereby partially offsetting the effect of bone loss. These beneficial changes include continued periosteal apposition, which increases cross-sectional area over life by about 15% in both sexes; this leads to outward displacement of the cortex and increases the moment-of-inertia and, thus, bone strength. This helps offset the decrease in bone strength caused by thinning of the cortex due to a greater rate of endosteal bone resorption than of periosteal apposition with aging. Using DXA, Duan et al. reported that the increase in cross-sectional area of the femoral neck was more in males than in females. However, DXA measurements are subject to volume averaging artifacts. Using QCT, in which this is less of a problem, it has been found that age-related increases in femoral neck cross-sectional area were similar in females and males .
32.2.3.2
Trabecular bone loss in premenopausal women
Most reviews and textbooks state that little, if any, bone is lost in women until after menopause and later in life in men. Although the possibility of earlier bone loss had been suggested by some longitudinal studies using DXA or cross-sectional studies using QCT, it is now clear that trabecular bone loss occurs in young adulthood. In a population-based, longitudinal study in which changes in vBMD were assessed by QCT at the lumbar spine, distal radius, and distal ulna, Riggs et al. found highly significant trabecular bone loss in young adult women between ages 21 and 50 years (or until menopause) that accounted for about one-third of their total trabecular bone loss over life. In contrast, cortical bone loss could not be detected in women until about 5 years before menopause, as had been previously reported using DXA by Slemenda et al. .
32.2.3.3
Accelerated transient phase of early postmenopausal bone loss
Menopause initiates a rapid phase of cortical bone loss and further accelerates the preexisting phase of trabecular bone loss (see Chapter 25 : Osteoporosis in men: what is similar and what is different?). This acceleration lasts for 4–8 years before decreasing back to a steady, continuous rate of loss. This process is most apparent after surgical menopause. Genant et al. made a 2-year longitudinal study on perimenopausal women undergoing elective ovariectomy who had been randomly assigned to groups receiving placebo treatment or E replacement therapy. As compared with the placebo group, trabecular bone loss from the vertebral centrum was 7%–9% as assessed by QCT, whereas cortical bone loss as assessed by single photon absorptiometry of the radius diaphysis or radiogrammetry of the metacarpals was only 1%–3%. Some of this apparent increase in bone loss early after menopause could have been due to expansion of the remodeling space associated with the well-documented menopausal increase in bone turnover . The rapid rate of trabecular bone loss detected by QCT is more difficult to demonstrate using DXA, which fails to distinguish between trabecular and cortical bone. The most definitive data using DXA were reported by Greendale et al. from the transnational, multiethnic SWAN cohort study of perimenopausal women followed for the 5 years before and 5 years after the final menstrual period. They estimated that the cumulative 10 year lumbar spine BMD loss was 10.6%, of which 7.38% was lost during the transmenopause (defined as from 1 year before through 2 years after the final menstrual period); the cumulative 10 year femur neck BMD loss was 9.1%, with 5.8% being lost during the transmenopause, findings similar to those reported for previous, smaller transmenopausal DXA studies . Longitudinal studies of bone loss for up to 16 years after menopause have been made only for the predominantly cortical bone of the peripheral skeleton . These more extended studies suggest that this rapid phase of cortical bone loss declines exponentially over 4–8 years to merge asymptotically into a subsequent slow phase of bone loss. It has been suggested that the termination of the accelerated postmenopausal stage may be mediated by a resetting of the mechanostat to maintain a level of lower bone mass in the presence of lower E levels . In trabecular bone the menopausal acceleration is additive to the premenopausal phase of trabecular bone loss, whereas the cortical bone loss begins at menopause or shortly before .
32.2.3.4
The late, slow phase of postmenopausal bone loss
The subsequent slow phase of bone loss becomes apparent after the postmenopausal accelerated phase subsides and differs from it in several ways . First, the rate of loss is slower, averaging about 0.5–2.0%/year, depending on the bone site measured and instrumentation used; however, in the very old with multiple comorbid conditions, the rate of bone loss appears to accelerate again. Second, in contrast to the preceding accelerated phase that occurs only in women or in gonadectomized men, the slow phase of bone loss occurs in both sexes. Third, in contrast to the accelerated early postmenopausal phase, which is transient, the slow phase continues indefinitely. Finally, although the early accelerated phase involves mainly trabecular bone loss, recent data indicate that the subsequent slow phase involves the loss of mainly cortical bone and increases in cortical porosity .
32.2.3.5
Changes in bone microstructure
It is clear that only about 3%–30% of the antifracture effect of anticatabolic (antiresorptive) drugs used to treat osteoporosis can be explained by treatment-induced increases in BMD and that the remainder is related to the prevention of microstructural damage caused by increased bone turnover . As previously reviewed , changes in bone microstructure of both trabecular and cortical bone can have profound effects on bone strength that are not captured by measurements using DXA or conventional QCT. These effects include the loss of trabecular cross-struts that disproportionately weaken bone, resorption cavities on the surface of trabecular bone that act as stress-risers to concentrate mechanical loads focally, and increased porosity of cortical bone. As originally demonstrated by Parfitt et al. using histomorphometry of biopsy samples and subsequently confirmed noninvasively by Khosla et al. using HRpQCT, the microarchitectural damage associated with loss of trabeculae occurs mainly in postmenopausal women, whereas changes in men are mainly due to trabecular thinning. Also, Ladinsky et al. used noninvasive magnetic resonance imaging to show that early postmenopausal women receiving placebo demonstrated progressive microarchitectural damage over a 1-year interval, whereas those treated with E were protected. This suggests that the high bone turnover initiated by menopause leads to microstructural damage that is independent of the concomitant decrease in vBMD. Indeed, as demonstrated by Silva and Gibson , loss of trabeculae decreases trabecular bone strength two- to fivefold more than a comparable decrease in BMD from age-related trabecular thinning.
Several studies have also highlighted the importance of increases in intracortical remodeling and in cortical porosity with aging . Increased intracortical remodeling with aging contributes substantially to increased cortical porosity and to a “trabecularization” of the endosteal surface . Interestingly, young and elderly women and men matched for aBMD by DXA at the ultradistal radius have similar trabecular parameters but the elderly subjects have marked increases in cortical porosity as assessed by HRpQCT; these changes in cortical microstructure with aging are not captured by DXA .
32.2.4
Patterns of change with aging in men
As assessed by cross-sectional and longitudinal studies using QCT, young adult (ages 20–50 years) men undergo an early phase of trabecular bone loss that is similar in time of onset and in rate of loss to that of premenopausal women and also does not involve significant losses of cortical bone ( Fig. 32.1 ). As assessed by longitudinal QCT measurements , cortical bone loss in men does not begin until about age 70 years, some two decades later than in women whose cortical bone loss is initiated at menopause. Moreover, although trabecular bone loss is continuous from young adulthood in men, there is an acceleration of this loss also at about age 70 years, at least at the lumbar spine. Because of continued periosteal apposition, however, the cross-sectional area of bone increases by about 15% over life in men.
32.3
Secretion and metabolism of sex steroids
As previously reviewed , the two major circulating forms of E are estradiol (E2) and estrone (E1); the former is about 3.5-fold more potent than the latter. In premenopausal women, >95% of serum E2 and most of serum E1 are derived from ovarian secretion. Peripheral conversion of C-19 steroid precursors, principally weak adrenal androgens, accounts for the remainder in premenopausal women and for almost all of the circulating E in postmenopausal women. Action of the enzyme, aromatase , which is found in many tissues but mainly in adipose tissue, is responsible for most of the extragonadal production of E in humans: E2 is derived from T, and E1 is derived from androstenedione. In addition to the extragonadal synthesis of circulating sex steroids, in some target cells, including osteoblasts, sex steroids are both synthesized and utilized intracellularly. Labrie termed this process “intracrinology” .
In men, T is the major potent circulating androgen, and >95% of it is derived from testicular secretion. In premenopausal women, 25% of serum T is derived from ovarian secretion, 25% from adrenal secretion, and 50% from peripheral conversion of weak adrenal androgens. The sources in postmenopausal women are similar except that ovarian secretion of T is less. In many target tissues, 5α-dihydrotestosterone (DHT), formed from T through the action of the enzyme 5α-reductase, is the principal androgen.
The circulating levels of active sex steroids are functions both of their rates of production and rates of removal. Although other pathways for steroid degradation exist, the two main ones for the removal of circulating E involve 2-hydroxylation and 16α-hydroxylation. The 2-hydroxylated estrogens are inactive or, in some experimental systems, antagonistic, whereas the 16-hydroxylated estrogens retain E activity . The major pathway for inactivation of circulating T is by oxidation to 17-ketosteroids.
Altogether, men make 20-fold more androgens than do women; the proportion of androgen converted to E2 is 200-fold more in women; and E2 is 1000-fold more potent than androgens (on a molar basis) on target tissues . Thus circulating E levels are measured in picograms, and T levels are measured in nanograms. These circulating levels can be partitioned into three components. The free steroid level, the freely available bioactive component, represents ~1%–3% of the total steroid. Another component (35%–55% of the total) is loosely bound to albumin and, because it is diffusible, also has biological activity. However, the largest component (40%–65%) is tightly bound to sex hormone-binding globulin (SHBG) and, thus, has limited biological activity. Because the level of SHBG may vary independently of total sex steroid levels, the non-SHBG bound fraction should be assessed to determine the clinical significance of sex steroid levels. This can be done by several methods. Moreover, the SHBG concentration is regulated by the sex steroids: E increases and androgens decrease SHBG.
32.4
Direct effects of sex steroids on bone
32.4.1
Effects at the cellular level
Bone resorption and bone formation do not occur randomly throughout the skeleton but are coupled together at discrete foci of functional assemblies termed basic multicellular units (BMUs). Over the course of a year, there are about 10 6 active foci, which follow the programmed temporal sequence of activation, resorption, and formation. At the cellular level, E suppresses the activation (birth rate) of BMUs and maintains a balance between the resorptive and formative phases. When E is deficient, the activation frequency of new BMUs increases and, at each of them, the resorptive phase is more than the formative phase. The enhanced resorption phase during E deficiency is mainly the result of prolongation of osteoclast life span due to inhibition of apoptosis , although an increase in osteoclast work potential may also contribute .
Although less well established than its effect on osteoclasts, some studies indicate that E increases osteoblast formation, differentiation, proliferation, and function . In addition, two groups have demonstrated that E antagonizes glucocorticoid-induced osteoblast apoptosis and, thus, extends osteoblast life span.
32.4.2
Transduction by sex steroid receptors
As with other steroid hormones, sex steroids act on target tissues by binding to specific nuclear receptors that act as transcription factors to activate specific genes. There are two species of estrogen receptors (ERs)—ERα and ERβ—but only a single androgen receptor (AR) species. Both ERs are found in osteoblasts: ERα is mainly found in cortical bone, and ERβ is mainly found in trabecular bone . ERα also has been demonstrated to be present in osteoclasts and osteocytes. AR is present in many tissues, including reproductive tissue, bone, and muscle. In reproductive and some other tissues, androgenic action is mediated by conversion of T to DHT. In addition, ERα and β as well as AR are also present in other cells in the bone microenvironment, such as T cells, which also regulate bone metabolism (as discussed later).
The altered configuration of the dimeric nuclear receptor following binding by sex steroids allows this complex to associate with various intracellular coregulator proteins (which can be either stimulatory or inhibitory). The complex then binds directly to specific sex steroid response elements on DNA or indirectly through protein–protein interactions to other DNA sequences such as the AP-1 or SP-1 sites. Increasing evidence also indicates that sex steroids may signal through rapid, membrane pathways involving regulation of specific kinases . Subtle changes in the configuration of the steroid nuclear receptor upon binding by different ligands allow association with a different complement of coregulator proteins. This association along with tissue differences in the relative concentrations and types of coregulator molecules are thought to be the major mechanisms responsible for differences in action by estrogens, antiestrogens, and selective ER modulators (SERMs) in different target tissues .
32.4.3
Effects on T cells and other immune cells
Significant evidence suggests that T cells and other immune cells play a pivotal role in postmenopausal bone loss. Much of the evidence derives from studies conducted in ovariectomized (ovx) mice. The field was opened by reports indicating that ovx fails to induce trabecular and cortical bone loss in T cell deficient nude mice . Confirmatory data came from the observation that protection against ovx-induced bone loss occurs in WT mice depleted of T cells by injection of anti–T cell antibodies , mice treated with Abatacept (an agent which blocks T cell costimulation and induces T cell anergy and apoptosis) and mice lacking the T cell costimulatory molecule CD40 ligand (CD40L) .
Both CD4+ and CD8+ cells play a role in ovx-induced bone loss. CD4+ cells include the Th1, Th2, and Th17 subsets. Th17 cells are an osteoclastogenic population of CD4+ cells defined by the capacity to produce IL-17 . Th17 cells potently induce osteoclastogenesis by secreting IL-17A, receptor activator of nuclear factor kappa-B ligand (RANKL), TNFα (TNF), IL-1 and IL-6, along with low levels of IFNγ . IL-17A stimulates the release of RANKL by all osteoblastic cells including osteocytes and increases the osteoclastogenic activity of RANKL by upregulating RANK . Th17 cells are first produced in the lamina propria of the small intestine. From there, they migrate to sites of inflammation, including the BM. Postmenopausal women with osteoporosis are known to have high levels of circulating IL-17 . In experimental animals ovariectomy promotes the differentiation of naïve CD4+ cells into mature Th17 cells, causing a net increase in the number of BM Th17 cells . Estrogen deficiency increases Th17 cell differentiation indirectly via increased production of cytokines such as TGFβ, IL-6, IL-1β, and TNF , factors which are all disregulated by estrogen deficiency . In addition, Th17 cell differentiation is inhibited by estrogen via a direct effect on CD4+ T cells mediated by ERα . The importance of IL-17 in bone loss is highlighted by the fact that silencing of IL-17R or treatment with anti–IL-17 antibody prevent ovx-induced bone loss.
Estrogen deficiency also influences regulatory T cells (Tregs), a population capable of suppressing the effector function of Th1, Th2, and Th17 T cells. Tregs are defined by the expression of the transcription factor FoxP3 and the ability to block inflammatory diseases and maintain immune homeostasis and tolerance . Tregs are comprised of thymus derived Tregs (tTregs, also known as nTregs) and peripherally derived Tregs (pTregs, also known as iTregs) . Defects in Treg numbers and/or activity have been implicated in several chronic inflammatory diseases. Tregs inhibit monocyte differentiation into osteoclasts in vitro and in vivo and blunt bone resorption through the secretion of IL-4, IL-10, and TGFβ1 . Attesting to the relevance of Tregs, E increases the relative number of Tregs . Moreover, transgenic mice overexpressing Tregs develop high bone mass as they age due to inhibition of bone resorption, and are protected against ovx-induced bone loss . Finally, adoptive transfer of Tregs into T cell deficient mice increases bone mass .
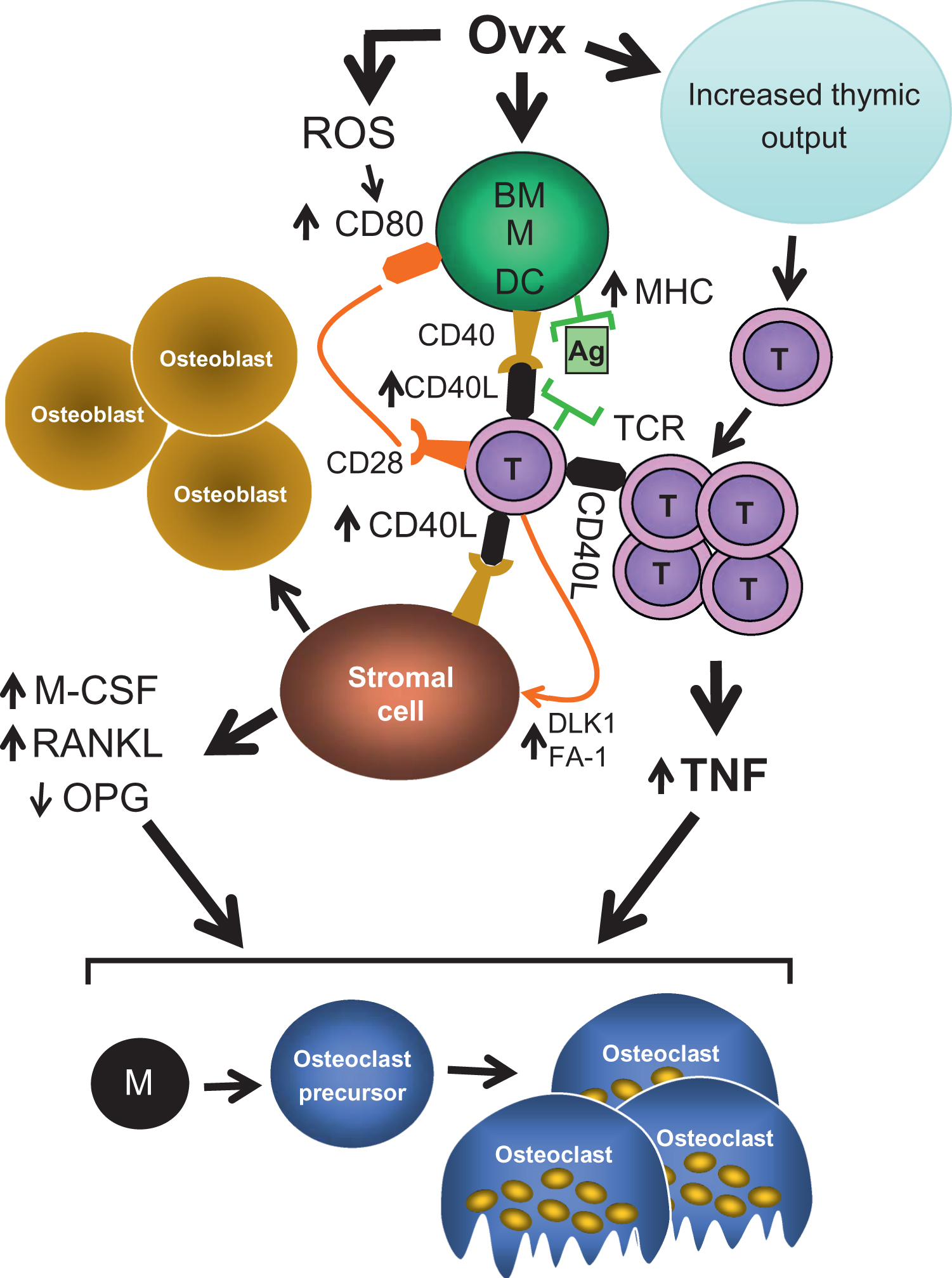
Two mechanisms have been described to explain how T cells contribute to ovx-induced bone loss ( Fig. 32.2 ). The first involves an increased production of TNF by BM T cells. The second is a regulatory cross talk between T cells and stromal cells (which include progenitors to multiple mesenchymal cell types, including osteoblasts, adipocytes, and chondrocytes) resulting in enhanced production of osteoclastogenic cytokines by the stromal cells.
Ovx increases T cell TNF production to a level sufficient to augment RANKL-induced osteoclastogenesis . This effect is due to an increased number of TNF producing T cells and enhanced the production of TNF per cell . Ovx also increases the population of prematurely senescent CD4+CD28− T cells , a lineage that produces high levels of TNF.
The presence of increased levels of TNF in the bone marrow of ovx’d animals is well documented . The ovx-induced increase in TNF levels is mostly due to T cell TNF production . Clinical evidence corroborates the role of T cell produced TNF in postmenopausal bone loss .
The relevance of TNF in ovx-induced bone loss has been demonstrated in multiple animal models. For example, ovx fails to induce bone loss in TNF knockout mice and in mice lacking the p55 TNF receptor . Likewise, transgenic mice insensitive to TNF due to the overexpression of a soluble TNF receptor , and mice treated with the TNF inhibitor TNF binding protein are protected from ovx-induced bone loss.
The mechanism by which E deficiency expands the pool of TNF producing T cells involves the reactivation of thymic function and induction of T cell activation in the BM ( Fig. 32.3 ). T cell activation is driven by enhanced antigen presentation by macrophages and dendritic cells (DCs) . The most upstream effects of ovx in the BM are to stimulate the production of reactive oxygen species (ROS) and to impair the generation of antioxidants . In response to ovx, ROS are produced by most BM cells, including T cells . ROS play an important role in postmenopausal bone loss by generating a more oxidized bone microenvironment . ROS have important direct effects on osteoblasts and osteoclasts that have been addressed elsewhere . However, additional pivotal effects of ROS include expanding the pool of mature DCs that express the costimulatory molecule CD80 and increasing the DC-mediated antigen presentation .

The question thus arises as to the nature of the antigens. Ovx induces T cell expression of activation markers and promotes T cell proliferation, expansion, and acquisition of effector functions. These are all features of T cells exposed to foreign antigens . The gastrointestinal tract is colonized for life with 100 trillion indigenous bacteria, creating a diverse ecosystem known as the microbiota, whose contributions to human health are profound . The microbiota is likely to represent the source of foreign antigens that drives the expansion of T cells induced by ovx . The role of the microbiota in the bone loss of estrogen deficiency is discussed in detail in Chapter 39 , Bone and the microbiome.
A second direct, upstream effect of E deficiency is to blunt BM levels of TGFβ , a powerful repressor of T cell activation. TGFβ acts as an immunosuppressant by inhibiting T cell activation and T cell production of inflammatory cytokines. Demonstrating the relevance of the repressive effects of TGFβ on T cell function, mice with T cell–specific blockade of TGFβ signaling were found to be completely resistant to the bone sparing effects of E . Gain of function experiments confirmed that elevation of the systemic levels of TGFβ prevents ovx-induced bone loss and bone turnover .
The key downstream mechanism by which ovx increases antigen presentation by macrophages is a stimulatory effect on the expression of the gene encoding Class II Transactivator ( CIITA ). The product of CIITA is a non-DNA binding factor induced by IFNγ that functions as a transcriptional coactivator at the MHC II promoter . Increased CIITA expression in macrophages derived from ovx mice results from ovx-mediated increases in both T cell interferon (IFN)γ production and the responsiveness of CIITA to IFNγ . This cytokine was initially described as an antiosteoclastogenic cytokine because it is a potent inhibitor of osteoclastogenesis in vitro . The notion that IFNγ is an inhibitor of bone resorption was reinforced by the finding that silencing of IFNγR signaling leads to a more rapid onset of collagen-induced arthritis and bone resorption as compared to WT controls, and by the report that IFNγ decreases serum calcium and osteoclastic bone resorption in nude mice . However, IFNγ is an effective treatment for osteopetrosis both in humans and rodents . This clearly demonstrates that the net effect of IFNγ in vivo is to stimulate osteoclastic bone resorption. In keeping with a net proresorptive effect of IFNγ in vivo, IFNγ−/− and IFN γ R−/−mice are protected against ovx-induced bone loss . Mice lacking IFNγ production are also protected against infection-induced alveolar bone loss , while in erosive tubercoloid leprosy and psoriatic arthritis IFNγ production correlates positively with tissue destruction . Finally, disruption of IFNγ signaling in vivo results in a strong and sustained inhibition of markers of osteoclastic activity . These opposing in vitro and in vivo effects of IFNγ are explained by the fact that IFNγ influences osteoclast formation both via direct and indirect effects . IFNγ directly blocks osteoclast formation through targeting of maturing osteoclasts . However, IFNγ is also a potent inducer of antigen presentation and thus of T cell activation. Therefore when IFNγ levels are increased in vivo, activated T cells secrete pro-osteoclastogenic factors and this activity offsets the antiosteoclastogenic effects of IFNγ . It should also be mentioned that it is now recognized that IFNγ affects bone turnover by promoting the commitment of marrow stromal cells into the osteoblastic lineage and their differentiation into mature osteoblasts . Accordingly, treatment with IFNγ reverses ovx-induced bone loss by promoting bone formation .
Another mechanism by which E regulates IFNγ and TNF production is by repressing the production of IL-7, a potent lymphopoietic cytokine and inducer of bone destruction in vivo . IL-7 receptor KO mice display increased bone volume and BMD . In contrast, IL-7 transgenic mice have expanded bone marrow cavities with focal osteolysis of cortical bone and eroded bone surfaces . IL-7 induces the production of RANKL by human T cells , and the injection of IL-7 into mice in vivo induces bone destruction by inducing T cell production of RANKL and TNF . Importantly, levels of IL-7 are significantly elevated following ovx and in vivo IL-7 blockade using neutralizing antibodies is effective in preventing ovx-induced bone destruction by suppressing T cell expansion and TNF and IFNγ production .
32.4.4
T cell thymic output and bone loss
The thymus undergoes progressive structural and functional decline with age, coinciding with increased circulating sex-steroid levels at puberty . By middle age most parenchymal tissue is replaced by fat, and in both mice and humans, fewer T cells are produced and exported to secondary lymphoid organs. However, the thymus continues to generate new T cells even into old age . In fact, active lymphocytic thymic tissue has been documented in adults up to 107 years of age . Under severe T cell depletion secondary to HIV infection, chemotherapy or bone marrow transplant, an increase in thymic output (known as thymic rebound) becomes critical for long-term restoration of T cell homeostasis. For example, middle-aged women treated with autologous bone marrow transplants develop thymic hypertrophy and a resurgence of thymic T cell output which contributes to the restoration of a wide T cell repertoire , although the intensity of thymic rebound declines with age.
The mechanism driving thymic rebound is not completely understood, but one factor involved is IL-7 . Importantly, IL-7 alone is not sufficient to enhance thymopoiesis in young mice but plays a more relevant role in aged mice .
Both androgens and E have a profound suppressive effect on thymic function. Accordingly, castration reverses thymic atrophy and increases export of recent thymic emigrants to the periphery , while sex steroids inhibits thymus regeneration by promoting thymocyte apoptosis and an arrest of differentiation . Restoration of thymic function after castration occurs in young as well as in very old rodents .
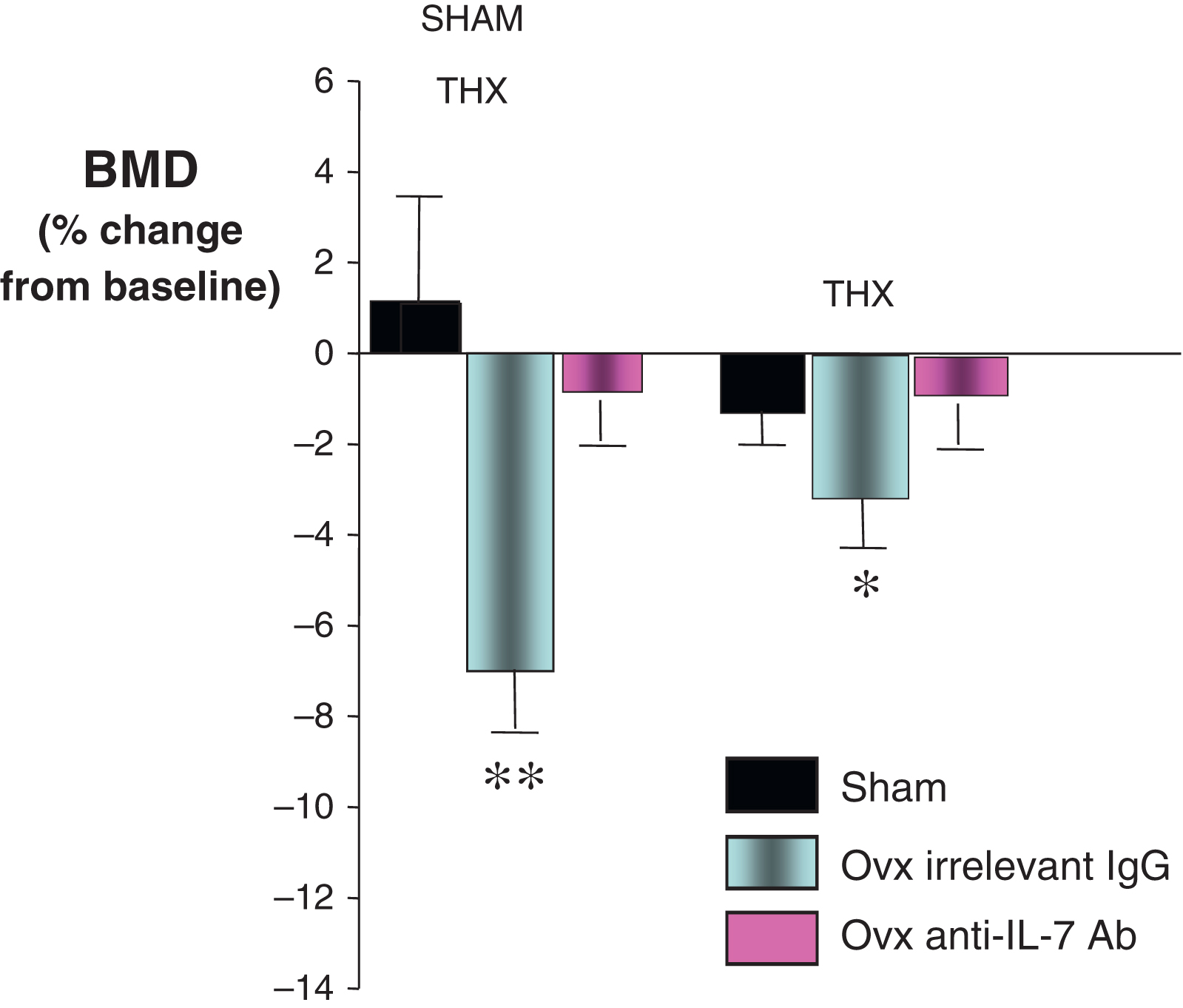
In accordance with the notion that E deficiency induces a rebound in thymic function, ovx expands thymic T cells and leads to the thymic export of naïve T cells . Indeed, stimulated thymic T cell output accounts for ~50% of the increase in the number of T cells in the periphery, while the remaining 50% is due to enhanced peripheral expansion. Similarly, thymectomy decreases by ~50% the bone loss induced by ovx ( Fig. 32.4 ), thus demonstrating that the thymus plays a previously unrecognized causal effect in ovx induced bone loss in mice. The remaining bone loss is a consequence of the peripheral expansion of naïve and memory T cells . This finding, which awaits confirmation in humans, suggests that E deficiency-induced thymic rebound may be responsible for the exaggerated bone loss in young women undergoing surgical menopause or for the rapid bone loss characteristic of women in their early postmenopausal period . Indeed, an age-related decrease in E deficiency-induced thymic rebound could mitigate the stimulatory effects of sex steroid deprivation and explain why the rate of bone loss in postmenopausal women diminishes as aging progresses .
32.4.5
Interactions between T cells and stromal cells
Several lines of evidence suggest that a cross talk between T cells and stromal cells is relevant for ovx-induced bone loss. First, activated T cells induce stromal cell apoptosis via the Fas/Fas ligand pathway, a phenomenon that blunts the compensatory increase in bone formation that limits bone loss in ovx mice . Second, a T cell/SC cross-talk driven by the CD40L/CD40 system is now known to regulate the production of osteoclastogenic cytokines by stromal cells and their osteoblastic progeny. CD40L is a key surface ligand expressed on T cells . CD40L binds to CD40 and several integrins . CD40 is expressed on antigen presenting cells, hemopoietic progenitors and cells of the osteoblastic lineage . CD40L has been linked to postnatal skeletal maturation because T cells, through the CD40L/CD40 system, promote the production of the antiosteoclastogenic factor OPG by B-cells . Consequently, CD40L deficient mice attain a reduced peak bone volume due to exaggerated bone resorption . Low bone density has also been found in children affected by X-linked hyper-IgM syndrome, a condition in which CD40L production is impaired due to a mutation in the CD40L gene . However, mice lacking T cell expressed CD40L are protected against parathyroid hormone (PTH) induced bone loss , raising the possibility that CD40L may exert antiresorptive activities in unstimulated conditions, while promoting bone resorption under conditions of bone stress.
Studies with CD40L null mice and with WT mice treated with the anti-CD40L Ab MR-1 have revealed that silencing of CD40L completely prevents ovx-induced bone loss . A dual mechanism was shown to be involved. First, silencing of CD40L blocks the activation of T cells and the resulting production of TNF. Second, CD40L is required for ovx to increase the proliferation and the differentiation of stromal cells and their capacity to support osteoclast formation through the enhanced production of M-CSF and RANKL and diminished the secretion of OPG. Thus a critical additional mechanism by which T cells dysregulate bone homeostasis in ovx mice is through a CD40L-mediated cross talk between T cells and stromal cells that results in enhanced osteoclastogenesis and, to a lesser degree, enhanced osteoblastogenesis.
In summary, ovx increases the number of activated CD40L expressing T cells that promote the expression of M-CSF and RANKL by SCs, and ovx downregulates the stromal cell production of OPG. The net result is a significant increase in the rate of osteoclastogenesis. The interaction of CD40L with CD40 on stromal cells in the context of E deficiency appears to override the protective effects of CD40/CD40L costimulation on basal B cell OPG production , distorting the balance of osteoclast formation in favor of bone loss.
An additional mechanism of T cells/stromal cell cross-talk involving delta-like 1/fetal antigen 1(DLK1/FA-1) has recently been described . Under physiologic conditions, DLK1/FA-1 is produced by stromal cells, B cells and T cells. These factors stimulate osteoclastogenesis and block osteoblastogenesis by inducing the production of TNF, IL-7, and other inflammatory cytokines by stromal cells. Ovx markedly increases the production of DLK1/FA-1 by activated CD4+ and CD8+ T cells, resulting in a further stimulation of the production of osteoclastogenic cytokines by stromal cells. Attesting to the relevance of DLK1/FA-1, DLK1 null mice are significantly protected against ovx-induced bone loss .
32.4.6
Effects on gasotrasmitters relevant to bone
H 2 S is a gasotransmitter released endogenously by mammalian cells. H 2 S is enzymatically generated in several tissues, including the vasculature, kidney, hearth, brain, nervous system, lung, upper and lower GI tract , and bone . H 2 S production is mainly controlled by two pyridoxal-5′-phosphate-dependent enzymes, cystathionine-β-synthase (CBS) and cystathionine-γ-lyase (CSE) . H 2 S acts a physiological messenger molecule in organs and tissues . First identified as a neuromodulator , H 2 S was found to be a potent vasodilator , to protect cells against oxidative stress by restoring glutathione levels and by inducing the nuclear translocation of Nrf2, the transcription factor that regulates a number of antioxidant genes . Moreover, H 2 S protect against inflammation by inhibiting lymphocytes infiltration in tissues and impairing T cell proliferation . H 2 S contributes to the acquisition and preservation of bone mass as it controls SC function by regulating Ca 2+ influx through Ca 2+ channels . H 2 S deficiency impairs the osteogenic differentiation of SCs .
Ovariectomy decreases serum H2S levels and the BM levels of CBS and CSE . Treatment with the H2 S-donor GYY4137 normalizes serum H2S in ovx mice, increases bone formation, and completely prevents the loss of trabecular bone induced by ovx . GYY4137 increases murine osteoblastogenesis by activating Wnt signaling through increased production of the Wnt ligands Wnt16, Wnt2b, Wnt6, and Wnt10b in the BM.
32.4.7
Effects at tissue and organ levels
As noted earlier, the main actions of E at the cellular level are to inhibit activation frequency of the BMUs and to maintain remodeling balance by inhibiting osteoclast function and, perhaps, by stimulating osteoblast function. At the tissue level, these cellular effects maintain equivalent levels of bone resorption and bone formation, and, at the organ level, this conserves bone mass . As assessed by calcium kinetics or biochemical markers of bone turnover, E deficiency leads to an increase in bone resorption. However, bone loss would not occur if the physiologic coupling between resorption and formation remained intact. Instead, in early postmenopausal women, bone formation increases only to about half of the level of bone resorption . Moreover, as assessed by three-dimensional bone histomorphometry, the impairment of bone formation in trabecular bone is even more severe in untreated patients with postmenopausal osteoporosis than in nonosteoporotic postmenopausal women, with a complete failure of osteoblastic compensation for increases in osteoclastic activity . Whether this failure of coupling is the result of E deficiency per se, age-related defects, or both is unclear.
The two most important physiologic mechanisms for maintaining bone mass are the action of E and the effects of mechanical strain. With few exceptions, such as states of corticosteroid excess, major decreases in bone mass do not occur unless one of these two homeostatic mechanisms is impaired. Frost hypothesized that strain is detected by an internal skeletal “mechanostat” that initiates changes in bone remodeling to adjust bone mass and distribution to a level that is appropriate for the ambient mechanical forces. He further hypothesized that E deficiency alters the set point of the mechanostat by decreasing the sensing of strain signals. Thus, if E is deficient, the mechanostat would erroneously sense lower strain interpreted as an excess of bone mass and initiate bone loss. In a series of studies Lanyon et al. demonstrated the molecular basis for this interaction. In ERα KO mice, Lee et al. demonstrated that the adaptive response of bone to mechanical loading is impaired, leading to an inadequate osteogenic response. Additional findings have shown that osteocytes are the major sensors of mechanical strain, that they contain ERα , and that E deficiency increases the concentration of ERα in osteocytes . The IGF-I receptor is also required in the efferent limb of the osteogenic response . These and other studies suggest that E action and the sensing of mechanical strain are functionally combined into a single system that acts to regulate bone mass. It should be noted, however, that more recent studies using deletion of ERα in the osteoblast lineage have, in fact, found that at least in female mice, loss of ERα leads to an enhanced (not reduced) response to mechanical strain . Thus additional studies are needed to more fully define the interaction of E, ER signaling, and mechanical loading.
The effects of T on bone at the tissue and organ levels are qualitatively similar to those of E, although much of this effect may be due to aromatization to E . Although it is unclear whether T is able to transduce the effects of mechanical strain as E does, it increases muscle mass and strength, which indirectly augment the stimulatory effect of weight bearing on bones .
32.5
Indirect effects of sex steroids on bone
32.5.1
Effects on peripheral calcium metabolism
It has been known for some time that E deficiency is associated with decreased intestinal calcium absorption and increased urinary calcium excretion and that both abnormalities could be corrected by E treatment. E increases intestinal calcium absorption both in experimental animals and in humans , acting through intestinal ER . E-responsive calcium influx channels have been shown to be present in intestinal mucosa . Also, E-deficient women have a blunted responsiveness to the action of active vitamin D metabolites that can be restored by E replacement . E also increases renal calcium conservation by enhancing tubular calcium resorption . In murine distal convoluted tubules, Oz et al. demonstrated that the molecular components required for calcium transport in wild-type mice were deficient in ArKO mice but could be induced by E treatment. Androgens also increase intestinal calcium absorption , although it is unclear whether they enhance renal calcium homeostasis as E does.
32.5.2
Direct effects on calcitropic hormones
Although E has direct actions on bone and peripheral calcium metabolism, some effects may be mediated indirectly by modulating the secretion of calcitropic hormones. Perhaps the best established of these is the ability of E to increase serum levels of 1,25-dihydroxyvitamin D (1,25[OH]2D). This has been difficult to demonstrate because oral administration of E increases the vitamin D binding protein by a “first pass” hepatic effect, thus spuriously elevating total 1,25(OH)2D levels. However, estimated free 1,25(OH)2D levels have been shown to increase after E administration or in pregnant women who have high endogenous E levels . Also, increases in calcium absorption in postmenopausal osteoporotic women following E administration correlated significantly with increases in serum total 1,25(OH) 2 D . Serum PTH decreases after E administration , and some have suggested that this results from a direct action on ER in the parathyroid glands. However, in detailed studies of basal and EDTA-stimulated PTH secretion in a group of untreated and E-treated early postmenopausal women, Vincent et al. failed to demonstrate an effect of E. A third calcitropic hormone whose secretion may be modulated by E is insulin-like growth hormone-I (IGF-I). However, several studies have shown that oral but not transdermal E decreases serum IGF-I . Since circulating IGF-I is mainly produced in the liver, this effect again may be a “first pass” artifact after oral administration.
32.6
Hormonal determinants of skeletal growth and maturation
Prior to puberty, basal levels of hormones from the growth hormone (GH)/IGF-I axis maintain slow, but continuous, bone growth. Puberty is initiated by increased pulsatile secretion of gonadotropin-releasing hormone (GnRH) by the hypothalamus, which leads to increases in serum gonadotropins, sex steroids, GH, and IGF-I . The increases in GH, IGF-I, and sex steroids act cooperatively to support the pubertal growth spurt. High pubertal levels of GH and IGF-I are maintained during the 3–4 years of rapid growth but then gradually decline to prepubertal levels over several years. In contrast, serum sex steroids increase to adult levels during puberty and then are maintained at that level . Despite never attaining supraphysiological levels, as IGF-I and GH do during puberty, the increase in serum E to adult levels is required for the pubertal growth spurt. Males with homozygous inactivating mutations in the ERα or the aromatase genes do not undergo the rapid pubertal growth spurt despite elevated serum levels of GH and IGF-I and normal or increased serum levels of T . Moreover, it is the continued rise in serum E levels during puberty that is the probable mechanism of epiphyseal closure in both sexes because young adult males who are unable to respond to E because of null mutations of the ERα or aromatase genes maintain open epiphyses, whereas men with testicular feminization due to null mutations of AR achieve epiphyseal closure . Thus E both initiates the pubertal growth spurt and then ends it by inducing epiphyseal closure.
Sex steroids also appear to increase bone mass during skeletal maturation independently of the effects of circulating levels of GH and IGF-I. The 25% greater bone mass in postpubertal boys over postpubertal girls is likely due mainly to the pubertal increase in serum T because increases in GH secretion and IGF-I production are similar or even greater in girls than in boys. E also contributes substantially to vBMD in both sexes. In a young adult male who was unable to synthesize E because of a null mutation in his aromatase genes, BMD was reduced by 25%–40% of predicted values at various skeletal scanning sites . However, in a study using pQCT in young postpubertal Swedish men, free T was found to be a positive and free E2 a negative predictor of cortical bone size, whereas neither cortical nor trabecular vBMD was associated with free T . Thus both T and E are major determinants of bone size and vBMD, although E appears to play the more important role, except for the long bones where stimulation of periosteal apposition by T is the main determinant of bone size .
32.7
Hormonal determinants of age-related bone loss in women
32.7.1
Accelerated phase of bone loss in early postmenopausal women
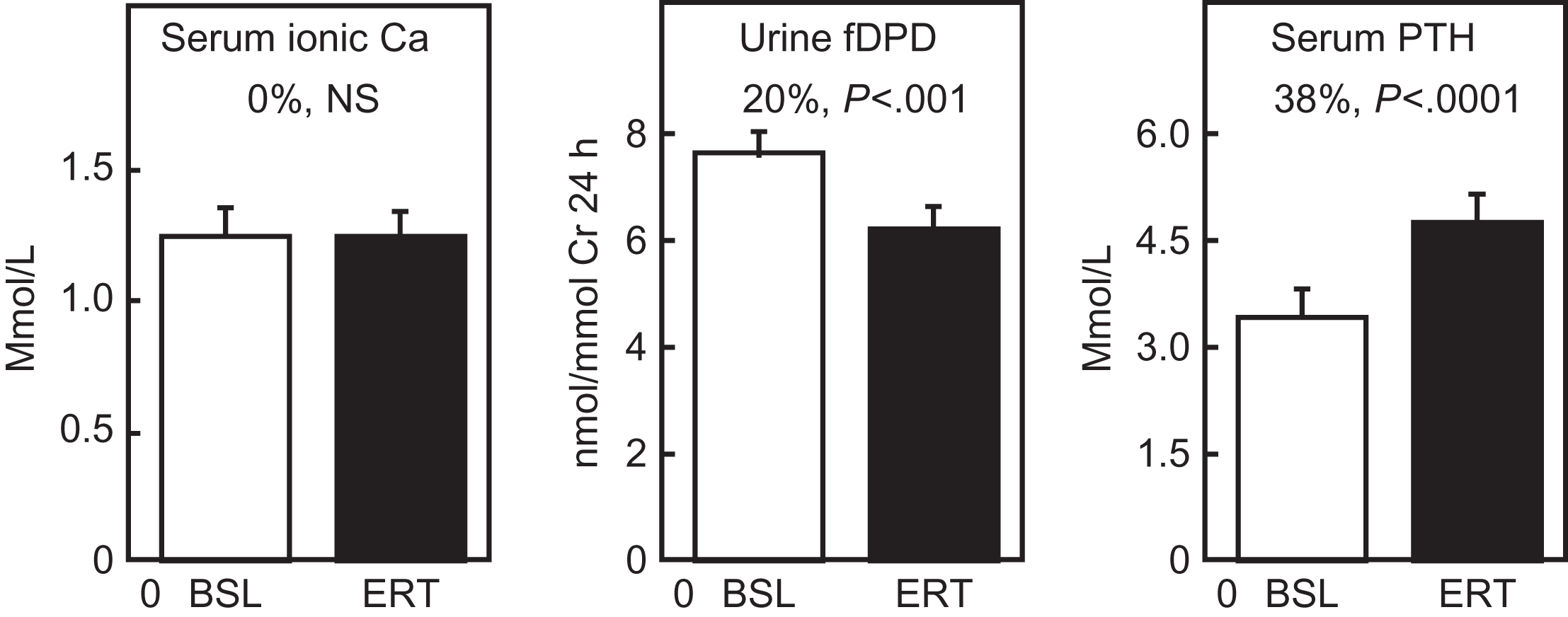
This phase begins at menopause, can be prevented by E replacement , and clearly results from loss of ovarian function (see Chapter 25 : Osteoporosis in men: what is similar and what is different?). During the 2- or 4-year menopausal transition, serum E2 levels fall to 10%–15% of premenopausal levels, although levels of serum E1, a fourfold weaker E, fall only to about 25%–35% of the premenopausal level . Serum T also decreases after menopause , but this decrease is only moderate because T continues to be produced by the adrenal cortex and by residual ovarian production. As assessed by biochemical markers, bone resorption increases by 90% at menopause, whereas bone formation markers increase by only 45% . The increase in bone turnover and the remodeling imbalance lead to accelerated bone loss, particularly from the endosteal surface of bone. The rapid bone loss in this phase produces an increased outflow of calcium from bone into the extracellular pool, but hypercalcemia is prevented by compensatory increases in urinary calcium excretion and decreases in intestinal calcium absorption , and by a partial suppression of PTH secretion . As shown in Fig. 32.5 , in the early postmenopausal period, serum ionized calcium is maintained at a constant level by a decrease in serum PTH, which compensates for the increase in bone resorption induced by E deficiency.