Estrogen and Progesterone Receptor Testing for Prognosis and Prediction
Mitchell Dowsett
William Miller
BACKGROUND
The major actions of steroid hormones are mediated through specific receptors that bind hormones with high affinity and thereby generate effective signaling. Some but not all breast cancers retain the hormonal sensitivity of the target organ in which they have developed such that their growth and development appear to depend on estrogen and, possibly, progesterone. As a consequence, treatments targeted at hormones and their signaling pathways have been used both to prevent and treat breast cancer. The hormonal sensitivity and therapeutic effects appear to be mediated through estrogen receptors (ERs) and progesterone receptors (PgRs). Consequently, measurement of these steroid receptors in breast cancers is used for estimating patient prognosis, particularly the likelihood of tumor response to and patient benefit from endocrine therapy.
Thus, the objectives of this chapter are to describe (a) the different forms of ER and PgR and their biology; (b) the methodology used to measure steroid receptors; (c) the utility of ER and PgR in determining clinical outcome of patients with breast cancer; and (d) the current status of the receptors in predicting the likelihood of response to treatment and therefore the selection of specific therapies in individual patients.
BIOLOGY OF ESTROGEN RECEPTORS (ERs) AND PROGESTERONE RECEPTORS (PgRs)
Structure and Function
ER and PgR belong to a family of nuclear hormone receptors that function as transcription factors when bound to their respective ligands.
Two separate ER isoforms have substantial homology, ERα and ERβ, and are encoded by two separate genes, ESR1 and ESR2, respectively (Fig. 26-1). The precise cell-specific physiologic and pathophysiologic roles of ERβ in breast cancer are currently unclear. Also few definitive data support any clinical role for ERβ at present, and routine evaluation of ERβ is rarely performed. Therefore, in the absence of qualification, the term ER refers to the product of ESR1 (ERα) throughout this review. PgR also exists in two separate but highly homologous isoforms (PgR A and B), which have been shown to have different regulatory effects but so far have not been shown to have significantly different predictive value.
Human ERα, ERβ, and PgR share common structural and functional organization with a central DNA binding domain (DBD) and a carboxyl-terminal hormone binding domain
(HBD) (Fig. 26-1). Binding the hormone to its specific receptors activates the receptors and facilitates binding to response elements present in the promoter of responsive genes. Coregulatory proteins coordinately act to influence transcription of responsive genes and influence the nature of response.
(HBD) (Fig. 26-1). Binding the hormone to its specific receptors activates the receptors and facilitates binding to response elements present in the promoter of responsive genes. Coregulatory proteins coordinately act to influence transcription of responsive genes and influence the nature of response.
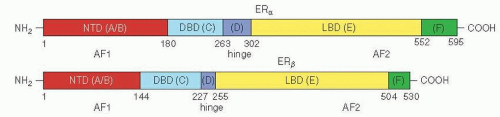 FIGURE 26-1 Linear organizational structure of ERα and ERβ. NTD: amino terminal domain; DBD: DNA binding domain; LBD: ligand binding domain; amino acid numbering for the extent of each domain is shown below the structure. (From Kumar R, Zakharov MN, Khan SH, et al. The dynamic structure of the estrogen receptor. J Amino Acids 2011;2011: 812540. Published online July 26, 2011.) |
Regulation
Mechanisms regulating ERα and β function include differential usage of upstream untranslated exons, the splicing of their messenger RNA (mRNA), and post-translational modifications (1). At least seven different promoters have been identified for ER (1). Alternative RNA splicing is relatively common in breast cancers, but there is little evidence that these result in equivalent protein variants that are detectable in clinical specimens, and none are recommended for use as prognostic or predictive tumor markers.
Post-Translational Modifications
Numerous post-translational modifications of ER and PgR have been reported, most notably through phosphorylation, ubiquination, and acetylation.
Phosphorylation of receptor protein is particularly influential. Several kinases can phosphorylate ERα, including p38 mitogen-activated protein kinase (MAPK), cyclin A-CDK2, CDK7, c-Src, and pp90rsk1 (2). Other important signaling molecules, such as AKT, and extracellular regulated kinase (ERK1/2) MAPK can also differentially phosphorylate ERα. Phosphorylation of ERα can occur at several sites and may alter response to ligands (2); for example, phosphorylation of ERα serine (S) 167 by AKT and S118 by ERK1/2 can produce ligand-independent activation of ERα (and thereby confer apparent hormone resistance) (3). ERα phosphorylation can also occur at S118, producing complex effects, and be decreased by endocrine therapy. Phosphorylation events are complex and interdependent and, for example, phosphorylation at ERα S305 can regulate the subsequent phosphorylation of S118 (4). Receptor phosphorylation also affects events such as receptor turnover, cellular localization, and transcriptional activity; however, the clinical utility of measuring ERα phosphorylation has not yet been demonstrated. Both PgR-A and B isoforms are phosphorylated at multiple serine residues (5), but how PgR serine phosphorylation regulates its function is not well defined.
Estrogen Receptor Gene Alterations
Only a few mutations have been reported in the ERα gene. The (K303R ERα) mutation that causes a single amino acid change in the ERα hinge domain leads to hypersensitivity to the growth effects of estrogen. One group has reported this to be present in about one-third of premalignant lesions and in one-half of invasive breast tumors (7). However, the literature is not consistent, and other studies employing different methodologies have failed to detect the mutation in invasive cancers (8), reported it in only 6% of breast cancers (9), or associated the mutation to a family history of breast cancer (10). Clearly, more definitive research needs to be performed.
Whether the ER gene locus (ESR1) is a target for increased gene copy number (amplification) is controversial and not as frequent as originally thought. However, amplifications between primary and metastatic tumors appear to be concordant, and tumors with ESR1 gene amplification also express higher levels of ERα by immunohistochemistry (11). Although some preliminary results suggest that ESR1 amplification may predict resistance to adjuvant tamoxifen in postmenopausal women with ER positive breast cancer (12), findings currently are not sufficiently robust to be used to define a subtype of primary breast cancers optimally suited for hormonal therapy on the basis of amplification. Further independent analyses of large series of breast cancers are warranted to determine the definite prevalence of ESR1 amplifications and its potential clinical significance.
Mechanism of Action: Genomic
ER and PgR function as tissue-specific and ligand-dependent transcription factors. Binding hormone to its receptor leads to a conformational change in the receptor and induces dimerization. The ligand/receptor complex then binds directly or indirectly to response elements in the promoter regions of responsive genes, enhancing transcription. The precise cellular response depends on tissue-specific nuclear coregulatory proteins, designated coactivators, and corepressors. More than 170 coregulatory proteins have been identified.
In the absence of hormone, histone deacetylase (HDAC) and receptor corepressors (such as N-CoR and SMRT) are bound to the receptor. Histone deacetylation silences or inhibits transcription by causing DNA to wrap more tightly around the core histone proteins. Once hormone binds to receptor, the activated complex displaces the repressor proteins, and acetyltransferases are recruited along with coactivator proteins (such as p160 coactivator, steroid receptor coactivator [SRC1], transcriptional inhibitory factor [TIF2], amplified in breast 1 [AIB1]) complex). The coactivators appear to cycle on and off the promoter during hormone treatment (13). There is therefore a dynamic and complex
array of proteins present on estrogen regulated promoters, many of which coordinately contribute to the hormonal regulation of gene expression.
array of proteins present on estrogen regulated promoters, many of which coordinately contribute to the hormonal regulation of gene expression.
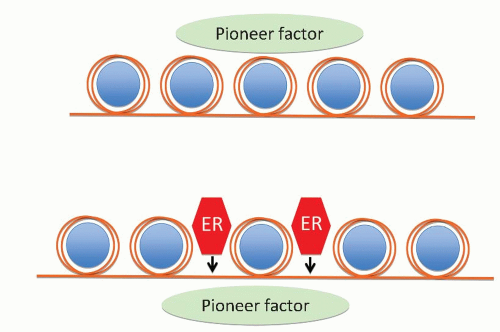 FIGURE 26-2 Mode of putative ER pioneer function. Upper panel: pioneer factor associates directly with compacted chromatin and provides accessibility to transcription factors such as ER to bind to DNA largely at oestrogen response elements. (Adapted from Jozwik KM, Carroll JS. Pioneer factors in hormonedependent cancers. Nature Reviews Cancer 2012;12:381-385.) |
Recent data indicate that the so-called pioneer factor FOXA1 cooperates with ER also to bind at large numbers of nonpromoter sites across the genomes (Fig. 26-2). The points through the genome that bind FOXA1 or ER vary among breast tumors and are affected by external influences such as growth factors (14).
Phosphorylation of ER coregulators is important in the transduction of signaling by the ER pathway. It can augment ER dependent transcription, even in the absence of ligand or in the presence of antiestrogens, by increasing subcellular nuclear localization and recruitment of other transcriptional coregulators to the receptor-promoter complex. Some coactivators, for example AIB1, are often gene-amplified or overexpressed in breast tumor cells. This may have clinical significance, and AIB overexpression has been associated with tamoxifen resistance, poor disease-free survival being observed after adjuvant tamoxifen therapy in patients whose tumors express high levels of both the ERBB2 oncogene, and the ER coactivator AIB1 (15). It may be the relative balance of bound coregulators that determines response to therapy.
Mechanism of Action: Nongenomic Activities
In addition to ER genomic activity in the nucleus, rapid effects of estrogens and plasma membrane estrogen binding sites have been described. It is possible that ER, PgR, and other steroid receptors can therefore mediate signaling cascades originating from the membrane or the cytoplasm through direct activation of signal transduction mediators. This nongenomic ER action occurs within seconds or minutes and is independent of gene transcription. Accruing evidence also suggests that such signaling may be associated with the growth and survival of breast cancer cells (16).
The identity of nongenomic receptors, their subcellular localization, and precise mechanism of action are still controversial and the topics of active research. However, immunohistochemical, biochemical, and genetic studies suggest that a subpopulation of the classic ERα and β subtypes located outside the nucleus and closely related nonclassic short forms of ERα may act as transducers of rapid estrogen signaling (17). These membrane and cytoplasmic ERs appear to transmit signals through kinase cascades, including growth factor receptors, cellular tyrosine kinases, and through calcium, cyclic adenosine monophosphate (cAMP), and other second messengers ultimately to regulate transcription in the nucleus (18). Membrane-initiated ER activity via growth factor signaling cascades can, in turn, modulate the activity of nuclear ER and its sensitivity to endocrine therapy (19).
In addition, nongenomic activity is also influenced by other cellular ER coregulatory proteins and by other pathways functioning in a given tumor. Increased expression of tyrosine kinase receptors (TKRs), such as in tumors amplified for HER2, can significantly augment ER nongenomic activity in response to both estrogen and tamoxifen (19).
Growth Factor and Estrogen Receptor Crosstalk-Implications for Hormone Resistance
Molecular bidirectional crosstalk occurs between growth factors, other signaling pathways, and the ER pathway. This crosstalk may be important in modulating ER activity and tumor response to endocrine therapies (20). For example, the bidirectional interaction between ER and the TKR pathway EGFR/HER2 can activate growth factor pathways by increasing the expression of ligands (i.e., transforming growth factor [TGF] α, amphiregulin), receptors (i.e., IGF-1), or other signaling intermediate molecules (e.g., insulin receptor substrate-1 [IRS-1]). Signaling through the HER pathway can also activate the transcriptional function of ER in the nucleus by phosphorylating coactivators and corepressors as well as ER itself (19).
There are also data that suggest that breast tumors with increased expression of growth factor signaling components, particularly of the EGFR/HER2 pathway, are associated with a poor response to tamoxifen (15, 21). Additionally, neoadjuvant trials observed higher response rates to aromatase inhibitors in HER2-overexpressing tumors as compared with those to tamoxifen (22, 23).
Although ER and HER receptors can amplify each other’s signals, inhibitory actions have also been observed. Activation of ER can down-regulate the expression of the HER receptor family, including EGFR1 and HER2, and HER signaling can down-regulate the expression of ER and PgR (24). It seems likely that the interaction between FOXA1 and ER in eliciting estrogen-dependent transcription is affected by phosphorylation of FOXA1, but the details of this remain to be elucidated.
Crosstalk raises the possibility that in some breast cancers, a simultaneous blockade of both ER and HER signaling pathways may be required to bypass resistance mechanisms and achieve optimal treatment benefit. Two recently reported randomized phase II trials comparing tamoxifen with or without gefitinib and anastrozole with or without gefitinib support this idea (25, 26).
OVERALL IMPORTANCE OF RECEPTORS IN CLINICAL BREAST CANCER
Approximately 30% to 40% of patients with ER positive metastatic disease responds to first-line hormonal therapies, and another 20% experience disease stabilization (27). Adjuvant hormonal therapy approximately halves the recurrence rate of patients with ER positive breast cancer. Hormonal therapy is also relatively nontoxic and therefore is a first-line option for virtually all patients with ER positive disease in both early and advanced disease. It is clear, particularly in the adjuvant setting (28) that patients with ER negative tumors do not derive benefit from endocrine treatment. Thus, ER acts as both a target and a biomarker for endocrine treatment.
The ER pathway can be targeted either by strategies that act on the receptor itself (i.e., selective ER modulators, such as tamoxifen, or potent pure antagonists that can degrade the receptor, such as fulvestrant) and by approaches that deprive the receptor of estrogen (i.e., aromatase inhibition and ovarian ablation). PgR is generally measured as a marker of an intact oestrogen-responsive pathway, and in the metastatic setting, it can aid in predicting a greater or lesser chance of response. However, in early breast cancer, PgR is helpful as a prognostic but not predictive marker of endocrine treatment benefit.
Before considering the importance of ER and PgR in breast cancer in more detail, it is instructive to understand the methodologies for their measurement in tissues.
METHODS FOR MEASURING ESTROGEN AND PROGESTERONE RECEPTORS
Assessment of ER status should be undertaken in all invasive breast cancers. Measurement of PgR is less important for selecting patients for endocrine therapy given that benefit is similar in ER+ PgR- and ER+ PgR+ cases. However, it is helpful in identifying the small population of ER- PgR+ tumors that merit endocrine therapy, and the identification can act as a quality control for ER measurement. Although ER status provides prognostic information, this is secondary to its value to assess the likelihood that a patient will respond to hormonal therapies.
Early studies relied on radiolabeled ligand-binding assays, such as the dextran-coated charcoal (DCC) method, which was rigorously validated and standardized in the United States. These methods were replaced in the 1990s with immunohistochemical (IHC) assays, which until recently have been subject to lower levels of QA.
Dextran-Coated Charcoal Ligand-Binding Assay (DCC-LBA)
The first assays of ER in breast cancer were introduced in the mid-1970s and were performed on crude tumor cytosols derived by centrifugation after homogenization. Tumor cytosols were incubated with high specific-activity radiolabeled steroid (estrogen or progestin), and the results reported as femtomoles (fmol) of receptor protein per milligram (mg) of total cytosol protein having been calculated from Scatchard plots in most instances (29). Although not used today, an understanding of the DCC assay is important because the data relating clinical benefit from endocrine therapy have been derived almost exclusively using this assay. The most widely used definition of positivity was at least 10 fmol/mg protein, but some described levels of more than 3 to 9 as borderline positive and negative as less than 3. Several disadvantages of the DCC assay existed, including variable tumor cellularity and heterogeneity as well as the requirement for fresh or snap-frozen tissue. These assays provide an overall score for the entire fragment of the tumor including neoplastic and non-neoplastic cells and may give false results, depending on the relative proportion of cancer versus other cell types within the tumor. Breast cancers display a broad dynamic range in ER and PgR expression using these assays. Overview analysis of 10,000s of patients treated with adjuvant tamoxifen showed little or no benefit for tumors with less than 10 fmol ER/mg protein, yet recurrence was reduced by about one-third in patients with tumors with 10 to 19 fmol/mg and by about one-half in those ≥200 fmol/mg. Mammographic screening dramatically reduced the average size of breast cancer below that required for the DCC assay. This and the availability of specific antibodies to ER and PgR led to the DCC no longer being performed for clinical management.
Immunohistochemical Assays
The development of specific, reliable, and commercially available ER and PgR antibodies (30) allowed the development of robust IHC technologies, and these are now virtually the only assays used to measure receptor levels. IHC allows for the determination of receptor status at the individual cell level, accommodating the problem of tissue heterogeneity within the tumor. IHC assays are less labor intensive and less expensive than extraction assays. They are also amenable to small tumors, and importantly, they can be performed on formalin-fixed, paraffin-embedded tissue, including archival tissues. IHC is also not affected by bound ligand (an issue with the DCC assay in pre-or perimenopausal patients).
IHC is performed on thin sections of formalin-fixed tissue that are subject to one of a number of antigen retrieval methods. This is followed by incubation of the section with a primary antibody directed against ER or PgR. Then a number of secondary detection systems, such as the use of secondary antibodies that have been conjugated to an enzyme such as horseradish peroxidase, are applied. The sections can finally be counterstained and viewed microscopically. For both ER and PgR, the staining produces a predominantly nuclear stain. The analytical systems have become increasingly sensitive and have resulted in most tumors being either completely negative or high positives (31, 32). Several scoring systems have been developed and implemented. Examples of ER and PgR staining are shown in Figure 26-3.
Comparison of Assay Methods and Standardization
There have been very few assessments of the relationship between IHC staining levels and benefit from endocrine
therapy, but those that exist and others that assessed prognosis either in untreated or hormonally treated tumors show good, although not perfect, concordance. When hormone receptor status of tumors determined by IHC assay has been compared with that determined by extraction assays, discordances between 10% and 30% have been reported for both ER and PgR status (33, 34). In some cases, IHCs have been found to have superior ability to predict hormone response in patients (35). Regan et al. (36) reported that, for ER status, concordance between IHC and DCC assays was higher among postmenopausal women (88%) than among those who are premenopausal (81%), possibly because of the interference that can occur in the DCC with high premenopausal estrogen levels. In contrast, concordance for PgR status was marginally lower in postmenopausal patients (76% vs. 80% premenopausal).
therapy, but those that exist and others that assessed prognosis either in untreated or hormonally treated tumors show good, although not perfect, concordance. When hormone receptor status of tumors determined by IHC assay has been compared with that determined by extraction assays, discordances between 10% and 30% have been reported for both ER and PgR status (33, 34). In some cases, IHCs have been found to have superior ability to predict hormone response in patients (35). Regan et al. (36) reported that, for ER status, concordance between IHC and DCC assays was higher among postmenopausal women (88%) than among those who are premenopausal (81%), possibly because of the interference that can occur in the DCC with high premenopausal estrogen levels. In contrast, concordance for PgR status was marginally lower in postmenopausal patients (76% vs. 80% premenopausal).
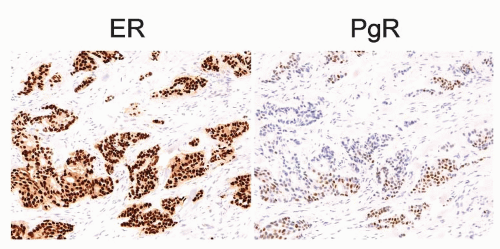 FIGURE 26-3 Examples of strong ER and weak PgR positive immunohistochemical staining in the same tumor. Brown nuclei are positively stained.
Related posts: Breast Cancer Screening Breast Cancer Screening
 Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations Ductal Carcinoma In Situ and Other Intraductal Lesions: Pathology, Immunohistochemistry, and Molecular Alterations
 Adjuvant Systemic Therapy: Endocrine Therapy Adjuvant Systemic Therapy: Endocrine Therapy
 Preoperative Endocrine Therapy for Operable Breast Cancer
Management Summary for the Care of Patients with Metastatic Breast Cancer
Nursing Care in Patient Management and Quality of Life Preoperative Endocrine Therapy for Operable Breast Cancer
Management Summary for the Care of Patients with Metastatic Breast Cancer
Nursing Care in Patient Management and Quality of Life
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|