Fig. 2.1
Chemical structures of DNA and RNA.
The DNA within the eukaryotic nucleus is arranged in a double-stranded helix composed of two strands of opposing polarity. The helix is stabilized by the formation of hydrogen bonds between complementary bases (A-T and G-C), by pi bonding that occurs when the bases are stacked together, and by the association of proteins [1, 2]. In eukaryotic cells, most of the DNA is in the B-form, a right-handed helix with bases on the inside where they are protected from damage by oxidating or alkylating agents. The Z-form of DNA occurs when a left-handed helix is formed, and is usually associated with portions of the DNA that are highly methylated and are not transcribed actively. Enzymatic reactions within the nucleus are responsible for conversion of DNA from the B-form to the Z-form and vice versa [3].
The DNA comprising the human genome is segmented into 46 discrete structural units, termed chromosomes. The DNA in each eukaryotic cell must be compressed to fit within the nucleus, which is only about 10 μm in diameter. Chromosomal DNA is condensed by the formation of nucleosomes, which consist of a group of small basic proteins (histones) with 160–180 base pairs of DNA wrapped around them [4]. Formation of nucleosomes is not sequence-dependent and it occurs in mammalian, bacterial, and viral DNA [5]. Nucleosomes are wound into a left-handed helix for further condensation of the DNA, and higher orders of structure include supercoils and/or rosettes [2]. Ultra-condensed DNA (heterochromatin) is inactive metabolically, and is found primarily in the periphery of the nucleus, while less condensed DNA (euchromatin) is readily accessible by transcription machinery and is located in the center of the nucleus [6].
2.1.2 Gene Organization
The majority of genes that are transcribed into mRNA and translated into cellular proteins exist as two copies in the nucleus of each cell, one maternal and one paternal copy. Some genes are present at a high copy number (100–250 copies) within the genome, including the genes that encode transfer RNA (tRNA), ribosomal RNA (rRNA), and the histone proteins [7]. These tandemly repeated genes are present on several chromosomes and associate in the nucleus to form a nucleolus [8, 9]. Highly repetitive sequences with thousands of copies, called satellite DNAs, are found at the telomeric ends of chromosomes and around the centromeres [10]. It is likely that the centromeric sequences play a role in the establishment and maintenance of chromosome structure. Telomeric repeats are involved in completing replication of chromosome ends [11], and it has been demonstrated that the length of the telomeric repeat sequences decreases with life-span of cultured human cells [12]. The evidence of telomeric shortening in normal cells along with observations that immortalized or transformed cells display limited telomere degeneration has led to the hypothesis that telomeric shortening is involved in the cellular aging process [12]. Other repetitive sequences, such as the Alu sequences, are found throughout chromosomes; their function is largely unknown, but a role in regulation of gene function has been proposed [13].
Simple polymorphic repetitive elements composed of dinucleotide or trinucleotide repeats are present in the human genome and have been associated with cancer and other diseases such as myotonic dystrophy, Fragile X syndrome, Huntington disease, and spinocerebellar ataxia [13, 14]. Symptomatic problems arise when the affected DNA is inappropriately methylated and inactivated (as in Fragile X syndrome) [15], or when the repeats cause detrimental changes in the encoded protein (as in Huntington disease) [16].
2.1.3 DNA Replication and Cell Division
In order for the DNA in a cell to be replicated prior to cell division it must be single-stranded. This is accomplished by an enzyme (helicase), which denatures the DNA and allows DNA-binding proteins to associate with the DNA and prevent reformation of the DNA helix [17]. A small strand of RNA 10–20 nucleotides in length acts as a primer, initiating synthesis of new complementary strands of DNA from multiple replication starting points. Deoxynucleotide triphosphates (dNTPs) are added to the primer by DNA polymerase, the RNA primers are removed, the gaps are filled in with dNTPs by DNA polymerase, and the nucleotide strands are joined by DNA ligase. The enzymatic action of topoisomerase removes twists generated during denaturation of the helix and allows the helix to re-form [18]. DNA replication is complete when the telomerase enzyme has added the nucleotide repeats to the telomeres at the 5′ end of the DNA strands.
As a new strand of DNA is synthesized, dNTPs are selected based on hydrogen bonding to complementary dNTPs in the template strand, which results in an error rate of 1 in 104–105 [2]. Eukaryotic cells employ a proofreading mechanism that removes mispaired dNTPs in the strand before the next dNTP is added, which decreases the error rate to 1 in 106–107 [2, 19]. Prior to cell division, another error correction system recognizes and repairs mismatched nucleotides and decreases the error rate further to 1 in 108–109 [2, 19]. Several inherited disorders are due to dysfunctional DNA damage-repair, including ataxia telangiectasia, Fanconi anemia, and xeroderma pigmentosum [20].
The DNA within the nucleus of a eukaryotic cell can be replicated completely in about 8 h, during the S phase of the cell cycle. Resting cells (G0) receive a mitotic stimulus, which causes transition into the G1 phase, where the cell prepares for DNA synthesis (S phase). The G2 phase occurs after replication but before division, and mitosis (M) involves actual nuclear and cellular division. The cell cycle is pivotal in cellular and organismal homeostasis, so it is tightly controlled by phosphorylation and dephosphorylation of kinases and cyclins, and by two major checkpoints [21, 22]. The first checkpoint occurs between G1 and S, and can prevent cells with damaged or unrepaired DNA from entering S phase. The second checkpoint occurs between G2 and M, and can prevent the initiation of mitosis [21].
2.1.4 Structure and Composition of RNA
RNA, or ribonucleic acid, is a linear polymer of ribonucleotides linked by 5′–3′ phosphodiester bonds (Fig. 2.1). RNA differs from DNA in that the sugar group of RNA is ribose, rather than deoxyribose, thymine is replaced by uracil (U) as one of the four bases, and RNA molecules are usually single-stranded. The extra hydroxyl group present on the ribose causes RNA to be more susceptible to degradation by nucleases than DNA. Single-stranded RNA molecules form complex secondary structures, such as hairpin stems and loops, via Watson–Crick base pairing between adenine and uracil, and between guanine and cytosine.
RNA molecules are classified by function and cellular location, and there are three major forms of RNA in eukaryotic cells. Ribosomal RNA (rRNA) is the most stable and most abundant RNA. rRNA is highly methylated, and complexes with proteins to form ribosomes upon which proteins are synthesized [23]. In eukaryotic cells, two major species of rRNA are present, 28S and 18S, as well as two minor species of 5.8S and 5S. Transfer RNA (tRNA) is responsible for carrying the amino acid residues that are added to a growing protein chain during protein synthesis. All tRNAs form secondary structures consisting of four stems and three loops, and many bases found in tRNA are modified by methylation, ethylation, thiolation, and acetylation [24, 25]. Messenger RNA (mRNA) mediates gene expression by carrying coding information from the DNA to the ribosomes, where the mRNA molecule is translated into protein. Messenger RNA is the most heterogeneous type of RNA, and also has the shortest half-life.
Many other RNA species exist in eukaryotic cells. Heterogeneous nuclear RNAs (hnRNA) are the precursors to mature mRNA [26]. Small nuclear RNAs (snRNAs) associate with proteins to form small nuclear ribonucleoprotein particles, which participate in RNA processing [27]. Small interfering RNAs (siRNAs) and microRNAs are double-stranded RNAs 21–25 nucleotides in length involved in regulation of gene expression at the translational level [28].
2.1.5 Transcription of RNA
Even simple eukaryotic organisms, such as yeast, contain a large number of genes (~2000), and higher eukaryotes, such as mammals, have ~20,000–25,000 protein encoding genes [29]. Clearly, the proteins encoded by all genes are not expressed simultaneously at any given time. Transfer of genetic information from DNA to protein begins with synthesis of RNA molecules from a DNA template by RNA polymerase, a process termed transcription. The RNA polymerase holoenzyme works processively, building an RNA chain with ribonucleoside triphosphates (ATP, GTP, CTP, UTP) [30]. Initiation of transcription involves association of the transcription machinery (RNA polymerase and transcription factors) with the DNA template and the synthesis of a small ribonucleotide primer from which the RNA strand will be polymerized [31]. Initiation of transcription is not random, but occurs at specific sequences called promoters that are located at the 5′-end of genes. Every gene initiates transcription independently at its own promoter, therefore the efficiency of the process varies greatly depending on the strength of the promoter. Once RNA polymerase binds to a promoter, the DNA helix is opened and an RNA primer is synthesized. Elongation occurs as the RNA polymerase moves along the DNA strand, opening the DNA helix and conducting DNA-directed RNA synthesis until the gene is transcribed [30]. Termination of transcription is poorly understood in eukaryotes, but takes place at sites that include a stretch of Ts on the non-template strand of the gene [32].
2.1.6 RNA Processing
The majority of eukaryotic RNAs require extensive modifications before they attain their mature structure and function. RNA strands may be modified by (1) the removal of RNA sequences, (2) the addition of RNA sequences, or (3) the covalent modification of specific bases. The long, relatively unstable mRNA precursor strand (hnRNA) is synthesized and remains in the nucleus where it is subjected to several stability-enhancing processes. With the exception of mitochondrial mRNAs, the 5′-ends of eukaryotic mRNA precursor molecules are capped, which involves removal of the terminal phosphate group of the 5′-nucleoside triphosphate and subsequent linkage of the 5′-diphosphate group to a GTP molecule [33]. The cap structure is covalently modified by methylation of the newly added guanine. The hnRNA is also modified at the 3′ end by the addition of a poly-A tail, a string of 50–250 adenine residues. The poly-A tail serves to extend the life of the mRNA by protecting the 3′-end of the molecule from 3′-exonucleases, and may also act as a translational enhancer [34]. The mRNA molecule is stabilized further by the association of a ~70 kDa protein with the poly-A tail [35].
The majority of protein-encoding eukaryotic genes contain intervening sequences, termed introns, which do not encode any portion of the protein. These introns, which may be between 65 and 10,000 nucleotides in length, are maintained during transcription of the hnRNA molecule, resulting in production of a long hnRNA that must be modified in order to become a continuous template for synthesis of the encoded protein. Maturation of the hnRNA requires a splicing event in which the introns are removed in conjunction with the joining of the coding sequences, termed exons. Three short consensus sequences are necessary for splicing to occur; two are found at the intron–exon boundary at both the 5′ and 3′ of the intron and the third is found within the intron near the 3′-end [36]. The consensus sequences mark splice sites and act as targets for the spliceosome, a large multisubunit protein complex comprised of 45 proteins and thousands of snRNA [36]. The spliceosome catalyzes removal of introns and rejoining of exons, ultimately resulting in the formation of a protein-encoding mRNA. Some genes encode for more than one protein, which is accomplished by alternative splicing of the primary hnRNA transcript. One mechanism of alternative splicing involves removal of one or more exons during splicing when the spliceosome ignores one or more intron–exon boundaries [37]. Alternative transcripts may also be generated by the use of a secondary polyadenylation site [38].
RNA processing is not limited to mRNA. Eukaryotic tRNAs are modified posttranscriptionally, as are eukaryotic rRNAs [39]. Introns present within rRNAs are classified as Group I or Group II introns. Group I introns are removed as linear molecules by a self-splicing mechanism that requires magnesium and guanosine as cofactors [40]. Group II introns are also self-splicing, are removed as a lariat structure, and require spermidine as a cofactor [41].
2.2 Basic Molecular Analysis and Interpretation
Investigations into the molecular mechanisms of disease depend on the analysis of cellular and often times microbial DNA and RNA to identify and characterize the genes involved. Target genes can be identified, localized to specific chromosomes, amplified by cloning, and subjected to sequence analysis. DNA analyses are used practically in the identification of individuals for forensic or parentage testing, detection of gene mutations, amplifications, and deletions, associated with disease and RNA analyses to characterize gene expression in various forms of human cancer.
2.2.1 Isolation of Nucleic Acids
In theory, nucleic acids can be isolated from any tissue that contains nucleated cells. Common starting material for nucleic acid isolations include blood and other body fluids, cultured cells, buccal swabs, and a variety of fresh, frozen, or formalin-fixed, paraffin-embedded (FFPE) tissue specimens. Although isolation may need to be optimized for any particular tissue type or sample and application, there are basic steps in the extraction process that are universal: the cells must be lysed, proteins and cellular debris must be removed, and the nucleic acid must be made available in solution.
FFPE tissue is the most common specimen type used when interrogating solid tumors for somatic changes in the clinical setting. FFPE tissue is readily available following tumor resection or biopsy, is easily stored at room temperature and the tissue being used for molecular analysis can be viewed microscopically to determine the precise histological diagnosis and to determine the adequacy of the specimen (percent tumor cells, exclude excessive necrosis, etc.). Nucleic acid derived from FFPE tissue, however, can be very problematic because the fixation process degrades nucleic acid and creates cross-links between nucleic acids and proteins. Molecular analysis of FFPE tissue, therefore, requires special attention during isolation and the design of downstream molecular applications. For example, the majority of an FFPE DNA sample is fragmented to 100-200 bp and will not work at all in most assays designed for high quality DNA. Assays specifically designed to detect these smaller fragments must be used in when FFPE-derived DNA is needed.
Before nucleic acids can be separated from the rest of the cellular contents, the cells must be lysed. This is often accomplished in some type of lysis buffer that can contain agents such as proteinase K and sodium dodecyl sulfate which are needed to digest structural proteins and to disrupt the cellular and nuclear membranes. Some samples such as solid tissues often require additional steps first, such as mincing, homogenization, or freezing the sample in liquid nitrogen followed by pulverization with a mortar and pestle to obtain a complete lysis. After the sample has been adequately lysed, the protein component in the sample must be removed. This can be accomplished in several ways, including: (1) adding a phenol–chloroform–isoamyl alcohol mixture to the sample which contains an organic phase and an aqueous phase whereby proteins and lipids dissolve into the organic phase and nucleic acids are retained in the aqueous phase of the mixture; (2) precipitation of proteins; (3) absorbance of nucleic acids to a column or magnetic bead and washing away proteins.
After the DNA has been separated from the rest of the other cell components, it must be transferred to a suitable buffer such as tris–EDTA (TE) or deionized water. This can be accomplished by adding ethanol or isopropanol which causes the DNA to precipitate out of solution. The DNA can then be pelleted by centrifugation, and resuspended in deionized water or a buffer. Alternatively, nucleic acids can be eluted directly from columns or magnetic beads in some of the more automated methods.
Due to the time involved and the toxicity and waste disposal issues associated with phenol, many laboratories choose other isolation protocols that remove proteins by other means. After lysis of the cells a protein precipitation solution can be added followed by centrifugation to separate the DNA from the cellular proteins. DNA can then be further purified by the use of silica gel particles in suspension or bound in a column. These silica columns can be purchased from numerous companies and are available in a wide range of formats and sizes, including the popular spin columns which rely on centrifugation to move solutions through the column. DNA binds to the silica gel as it passes through the column, allowing unwanted salts and contaminants to wash through. After the DNA is bound to the column it can be washed and then eluted off the column with water or a low salt buffer such as TE. Similar methods have been developed using magnetic beads capable of binding DNA in solution. DNA can be purified by binding it to these beads in a tube. When these tubes are placed next to a magnet, the beads can be held in place on the side of the tube while the original solution and contaminants are removed. This process can then be repeated with wash solutions and finally an elution buffer that is often heated to facilitate removal of the DNA from the beads.
Although these alternative methods can be faster, they are often more expensive and can result in lower yield and slightly lower quality DNA. However, this is not generally a problem for most molecular techniques commonly used today. Another improvement on traditional nucleic acid isolation methods has been the introduction of automated isolation systems which can greatly reduce the hands-on time needed for isolating DNA. Once loaded, these instruments can go through the entire DNA isolation procedure, often using some form of magnetic separation. Although these automated systems are much more expensive than manual methods, the time they save will often make these systems a valuable investment for laboratories that process larger numbers of samples.
RNA isolation methods are similar to those for DNA. However, when isolating RNA extra care must be taken to avoid RNA degradation due to its susceptibility to RNA-digesting enzymes (RNases). To avoid RNase digestion of the sample, it is often necessary to work quickly and to work in a clean area. Care should be taken to ensure that all solutions purchased for use in these extractions have been designated as “RNase-free” or treated with special chemicals such as diethylpyrocarbonate (DEPC) that inactivate RNases.
2.2.2 Use of Enzymes in Molecular Biological Techniques
DNA or RNA isolation techniques only produce the starting material needed for molecular evaluation of nucleic acid. Almost every method used to examine DNA or RNA relies heavily on the use of enzymes, often isolated from various microbial species. These enzymes can be exploited to perform various tasks such as selectively cutting DNA or RNA into smaller pieces or digesting them completely, modifying the ends of DNA strands to remove or add phosphate groups, ligating two strands of DNA together to form one, and replicating nucleic acids.
2.2.2.1 Nucleases
Nucleases make up a diverse group of enzymes that are able to cleave or digest nucleic acids. This group of enzymes can be divided into various classifications based on the type of nucleic acid they use as substrates and the way in which they digest. Exonucleases begin digesting nucleic acids at the ends while endonucleases make internal cuts in a nucleic acid molecule. Restriction endonucleases make up a very large group of enzymes found in bacteria that recognize very specific sequences of DNA, often six base pairs in length. For example, the restriction endonuclease EcoRI (isolated from E. coli) recognizes the sequence GAATTC and makes a single cut within this recognition site. If the sequence of a particular piece of DNA is unknown, observing the digestion patterns of various restriction enzymes can provide valuable information about its sequence. For example, if a particular mutation occurs within the recognition sequence of a specific restriction enzyme, that enzyme may not cut when the mutation is present.
When complete digestion of DNA or RNA is desired, for example when DNA is being isolated and contaminating RNA is not desirable, RNase can be used to selectively digest the RNA, leaving a pure population of DNA. In the same way , DNases can be used to digest unwanted DNA molecules in an isolation of RNA.
2.2.2.2 DNA Ligases, Kinases, and Phosphatases
In addition to digesting DNA, enzymes are also available which facilitate the joining or ligation of two or more DNA fragments. Enzymes with this capability are called DNA ligases and commercially, T4 DNA ligase is widely available and most often used in molecular laboratories. Since DNA ligases work by creating a phosphodiester bond between the 5′ phosphate and 3′ hydroxyl termini of two DNA strands, the presence of the 5′ phosphate is essential. DNA phosphatases, such as calf intestinal phosphatase or shrimp alkaline phosphatase, remove the phosphate group on the 5′ end of a DNA strand, preventing ligation; a DNA kinase, T4 polynucleotide kinase, can restore the 5′ phosphate in order to allow ligation.
2.2.2.3 Polymerases
DNA and RNA polymerases are enzymes that synthesize new strands of DNA or RNA, normally using a preexisting strand of DNA (or RNA) as a template. Reverse transcriptase is another polymerase that is unique in its ability to use RNA as a template for synthesizing DNA. DNA polymerases are essential for the polymerase chain reaction (PCR), a technique described below that has revolutionized molecular biology.
2.2.3 Electrophoretic Separation of Nucleic Acids
Gel electrophoresis of DNA is widely used in procedures such as Southern blotting and PCR in which separation or sizing of a population of DNA fragments is an essential step in the analytical process. In some cases, a significant amount of information can be gleaned from relatively simple electrophoretic procedures that take advantage of the various properties of DNA molecules (i.e., charge, size, and conformation). Gels employed in electrophoretic techniques are generally composed of agarose or polyacrylamide, which act as a matrix through which DNA (or RNA) will travel when an electric current is passed through the gel. Generally, larger fragments of DNA will migrate more slowly through the gel and smaller fragments will migrate more quickly. DNA of a given size will travel through the gel at the same rate and form a band that is visualized by staining the gel with a dye such as ethidium bromide, which binds to DNA and fluoresces when viewed under UV light (Fig. 2.2). Agarose gels are useful in most traditional molecular techniques for visualizing DNA greater than 100 base pairs. However, when smaller DNA fragments are to be analyzed, special agarose formulations or polyacrylamide gels are needed for proper resolution and visualization.
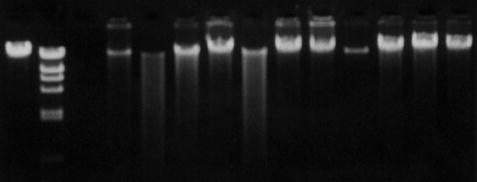
Fig. 2.2
Agarose gel electrophoresis of human genomic DNA.
When larger fragments of DNA need to be resolved, a technique called pulsed-field gel electrophoresis can be utilized. Conventional gel electrophoresis techniques are not useful for separation of extremely long pieces of DNA, because the constant current eventually unravels the DNA strands completely so that they travel, end first, through the gel at a rate that is independent of their length. Pulsed-field gel electrophoresis (PFGE) overcomes this challenge by periodically switching the orientation of the electric fields with respect to the gel, thus preventing the DNA strands from losing secondary structure and allowing long strands to be size-differentiated [42]. PFGE is often used in the identification of pathogens [43], i.e., differentiating between strains of bacteria (Fig. 2.3). Other applications include chromosomal length polymorphism analysis, and large-scale restriction and deletion mapping in DNA that is hundreds or thousands of kb in length. The effectiveness of PFGE is dependent on high-integrity starting material, and DNA that is degraded or sheared will not yield informative results. High quality DNA is often generated by embedding the cells of interest in agarose plugs, lysing cell membranes with detergent, and removing proteins enzymatically, leaving intact DNA which can be digested by restriction enzymes in situ and easily loaded into a gel apparatus [42, 44]. In a typical analysis, PFGE will produce a pattern of DNA fragments that range in size from 10 to 800 kb.
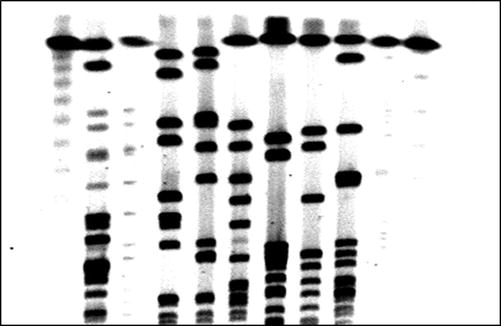
Fig. 2.3
Pulse field gel electrophoresis for strain typing of Staphylococcus aureus.
Electrophoresis of RNA to check the quality of a sample or for blotting techniques is typically accomplished by electrophoresis through a 1–2 % agarose gel containing formaldehyde, which maintains the denatured state of the RNA strands. When secondary structure of an RNA molecule is to be investigated, samples are subjected to non-denaturing polyacrylamide gel electrophoresis.
2.2.4 Denaturation and Hybridization
The majority of techniques used in molecular analyses require single-stranded nucleic acids as starting material. Prior to hybridization with a complementary nucleotide sequence such as an oligonucleotide primer or a nucleic acid probe, double-stranded DNAs and single-stranded RNAs must be denatured to generate single strands and eliminate secondary structure. Denaturation of nucleic acids is rapid, and can be induced by various conditions, including extremes of pH (pH < 4 or pH > 10), hydrogen-bond disrupting agents (such as urea or formamide), or heat [45]. Heat is the most commonly used means of disrupting the hydrogen bonds that occur between complementary strands of double-stranded DNA to produce single-stranded DNA. The melting temperature (T m) of a specific double-stranded DNA sequence is reached when the disruption of the hydrogen bonding in a population of DNA causes 50 % of the DNA to become single-stranded. Since more hydrogen bonding occurs between G and C as compared to A and T, the T m will be greater for DNA containing a larger percentage of G and C nucleotides. The denaturation process is easily monitored by spectrophotometry, since the absorbance of the DNA at 260 nm increases as denaturation progresses [45].
Hybridization of nucleic acid strands is a relatively slow process and the rate is governed by the relative concentration of strands with complementary sequences and by the temperature. When two complementary strands are aligned properly, hydrogen bonds form between the opposing complementary bases and the strands are joined. Target nucleic acid sequences can hybridize to a complementary DNA (cDNA) or RNA strand, or to other complementary sequences such as oligonucleotide primers or nucleic acid probes. Hybridization between two complementary RNA molecules is strongest, followed by RNA–DNA hybrids, and DNA–DNA hybrids [46].
2.2.5 Concepts and Applications of Southern Blotting
Southern blotting is a technique that relies heavily on the concepts of denaturation and hybridization of DNA. Southern blotting has been used in clinical or forensic settings to identify individuals, determine relatedness, and to detect genes associated with genetic abnormalities or viral infections. Southern blot analysis is also used in basic scientific research to confirm the presence of an exogenous gene, evaluate gene copy number, or to identify genetic aberrations in models of disease. Although this technique has been replaced to a large extent by newer, faster techniques, it still has its place in a molecular laboratory.
The first step in successful Southern blotting is to obtain DNA that is reasonably intact (Fig. 2.4). DNA that has been degraded by excessive exposure to the elements or mishandling will not produce a good quality Southern blot because it cannot be fragmented uniformly prior to the blotting procedure. The test DNA must be fragmented with restriction enzymes, which cut the double strands of DNA at multiple sequence-specific sites, creating a set of fragments of specific sizes which represent the regions of DNA between restriction sites. The fragmented DNAs are size-fractionated via agarose gel electrophoresis and are subsequently denatured, which enables them to later be hybridized to complementary nucleic acid probes. The DNA from the gel is transferred to a solid support such as a nylon or nitrocellulose membrane via capillary action or electrophoretic transfer and is bound permanently to the membrane by brief UV crosslinking or by prolonged incubation at 80 °C. Blots at this stage may be stored for later use or may be probed immediately. For detailed protocols, the reader is referred to other sources [46, 47].
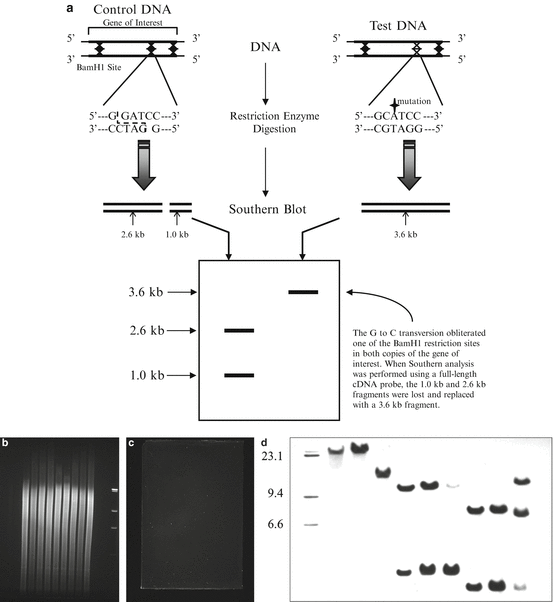
Fig. 2.4
Southern blotting. Variations in DNA sequences that alter restriction endonuclease recognition sites can be detected by Southern blotting (a). After digesting genomic DNA with restriction endonucleases, the DNA is subjected to agarose gel electrophoresis and then transferred to a membrane. The agarose gel can be imaged before (b) and after (c) the transfer has taken place to confirm adequate amounts of digested DNA and proper transfer of the DNA from the gel to the membrane. The membrane is then incubated with radiolabeled or chemiluminescent DNA probes which hybridize to specific DNA fragments. Exposing the membrane to film allows for visualization and sizing of these fragments (d).
Interpretation of the Southern blot is easy when the question is whether or not a gene is present in a particular sample, as long as appropriate positive and negative controls are included (a sample of DNA known to be positive and a sample of DNA known to be negative). The presence of multiple copies of a gene indicates gene amplification, which may occur in oncogenes during cancer development [48, 49]. Amplifications are obvious on a Southern blot as a band or bands that are more intense than the normal single-copy control; numerical values that reflect intensity may be assigned to bands using a densitometer. Structural aberrations in a gene of interest can be detected by Southern blotting, including the insertion or deletion of nucleotides or gene rearrangements (Fig. 2.4). When nucleotides are mutated, inserted, or deleted, the ladder of fragments produced may be abnormal due to the obliteration of restriction sites, the generation of novel restriction sites, or alterations in fragment size due to an increase or decrease in the number of nucleotides between restriction sites. The majority of these aberrations are apparent on Southern blots as abnormal banding patterns. The Southern blot remains a useful and reliable way to obtain definitive data on gene structure.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


