Chapter 51 Epstein–Barr Virus and Kaposi Sarcoma-Associated Herpesvirus
Epstein–Barr virus (EBV), the agent of infectious mononucleosis, is associated with the HIV-related syndromes of oral hairy leukoplakia (OHL) and non-Hodkgin lymphoma (NHL), including primary CNS lymphoma and Burkitt-like anaplastic lymphomas. In other populations, it has been implicated in the pathogenesis of post-transplant lymphoproliferative disease, nasopharyngeal carcinoma, and some forms of Hodgkin’s disease. An infectious etiology was long suspected for Kaposi sarcoma (KS) based on epidemiologic evidence. The identification of a herpesvirus, closely related to EBV, in KS biopsies has revolutionized our understanding of this disease. Information on this virus, called human herpesvirus 8 or KS-associated herpesvirus (KSHV), has accumulated at an astounding rate. The complete sequence of the virus was published just 2 years after its discovery. KSHV has subsequently been linked to other forms of neoplasia: primary effusion lymphoma (PEL) and the multicentric form of Castleman’s disease (MCD). EBV and KSHV, like all herpesviruses, can switch to a latent form of infection in which most of the viral genes are not expressed and the genome is maintained in the nucleus by cellular machinery. It is well established that the EBV genes expressed during latent infection of B lymphocytes drive them to proliferate and are almost certainly responsible for EBV’s association with malignancy. The pathophysiology of KSHV-related diseases is less completely understood, but expression of viral genes during latency appears to play a prominent role. The natural history and pathophysiology of these two viruses and implications for therapy are discussed in this chapter.
TAXONOMY
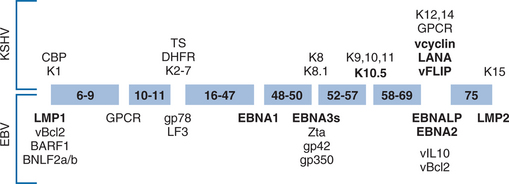
Human herpesviruses have diverged to fill unique biologic niches, but all employ the same basic replication mechanisms. Latent infection permits herpesviruses to persist quiescently in their hosts for decades and, from an evolutionary standpoint, has placed them among the most successful of human parasites. Five of the eight known human herpesviruses infect >90% of the population. Based on genomic organization and biologic characteristics, EBV and KSHV belong to the gammaherpesviridae subfamily of herpesviruses (Fig. 51-1). Their genomes are ∼170 kb in size and encode ∼90 proteins each. About 60 of these proteins are derived from the common ancestral gammaherpesvirus from which EBV and KSHV diverged ∼100 million years ago.1 A substantial fraction of the nonancestral proteins are expressed during latency (Fig. 51-2), underscoring the importance of latent infection for the adaptation of each virus to its biologic niche.
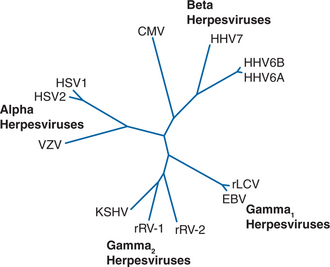
The two strains of EBV, type I and type II, show an overall divergence of <4%, and are indistinguishable on clinical grounds or by commercially available serologic tests.2 Infection with type I EBV is far more prevalent in most populations, although type II is more common among HIV-infected patients as is co-infection with type I and II. Five major subtypes of KSHV have been designated A–E.3,4 Unlike the situation with EBV, KSHV subtypes demonstrate distinct geographic distribution: A and C predominate in Europe with C extending to Asia and the Americas as well, B is predominately in Africa, and D and E are restricted to Oceania or Amerindian populations.4,5 It is not currently known if these subtypes differ significantly in their biologic properties.
EBV
Epidemiology
Seroepidemiologic studies show >95% of the adults are infected with EBV. In developing countries, primary infection occurs in infancy or early childhood, but in affluent populations of industrial countries, as many as one third of infections occur in adolescence or early adulthood. With the exception of vertical HIV transmission, EBV infection precedes HIV infection. Studies in children who acquired HIV infection by vertical transmission reveal that EBV seroconversion, at least in this HIV-positive population, did not have appreciable effect on clinically important parameters such as CD4 count or viral load.6,7 Moreover, primary infection appears to be largely asymptomatic, although one study noted an increased incidence of hepatosplenomegaly. By contrast, many studies have shown significant increases in the frequency of oral shedding of EBV by HIV-positive persons.6,8,9 As previously stated, type II EBV infection, rare outside of Africa and New Guinea, is present in about half of the HIV-positive population and at least half of these are co-infected with type I and II. In nonendemic areas, acquisition of type II EBV infection appears to be closely linked to sexual behavior.10,11
Pathogenesis/Molecular Biology
Multiple factors probably account for the interaction of HIV and EBV infections. First, persons with AIDS have 10–20 times as many circulating EBV-infected B cells. T lymphocytes from these patients have been shown to suppress EBV-positive B cells less effectively than do T cells from HIV-negative controls.12 Increased salivary shedding of EBV in the HIV population indicates that control of lytic infection is also impaired. Host factors also appear to be important as evidenced by the observation that HIV-positive persons with the 39A allele of the stromal cell-derived growth factor 1 (SDF-1) chemokine have a twofold increased risk of NHL in the heterozygous state and a fourfold increase when the host is homozygous.13,14 The chemokine receptor 5 variant CCR5D32 and the chemokine receptor 2 variant CCR2-64I, which are protective against progression to AIDS, were investigated in one study. The study found that the CCR5D32 allele conferred a threefold lower risk of developing NHL, but carrying the CCR2-64I had no effect on NHL risk.13
EBV-associated disease is distinctive because it is largely caused by latent infection not by viral replication. OHL, caused by extensive EBV lytic replication in oral epithelium, represents a notable exception. Although viral particles are not produced, latent EBV infection of B lymphocytes is not passive. In fact, EBV expresses two membrane proteins (LMP1 and LMP2), six nuclear proteins (EBV nuclear antigens or EBNAs), two untranslated RNAs (EBERs), and multiple micro-RNAs during latency.15 It is these viral proteins that transform B lymphocytes into LCLs, and are almost certainly responsible for EBV’s association with malignancy. Genetic analysis and biochemical techniques have begun to define the mechanisms by which these viral genes work.16 LMP1 is the major viral oncogene and induces a growth promoting signal that mimics a constitutively active form of the B-cell surface molecule CD40. The nuclear protein EBNA-1 ensures that the viral episome is maintained by cell machinery. A second nuclear protein, EBNA-2 is a powerful activator of transcription which targets downstream elements of the Notch signaling pathway to induce expression of both viral and cellular genes, including c-myc. The function of the remaining four nuclear proteins is less completely understood, but they may modulate the effects of EBNA-2.
The expression of all 10 latent EBV genes, referred to as type III latency, induces a potent immune response and is only seen in the peripheral blood of normal hosts during infectious mononucleosis. After resolution of primary infection, more restricted latent gene expression is observed. Continued EBNA-1 expression assures maintenance of the viral genome and, because EBNA-1 can inhibit its own processing for presentation on class I MHC molecules, does not incite a significant immune response.17 The EBERs and LMP2 also continue to be expressed and, though they appear to have no direct role in lymphocyte transformation, they may be important for the biology of EBV in vivo. For example, LMP2 has been shown to interact with signaling proteins downstream of the B cell receptor to prevent B cell activation. Since this signal frequently induces lytic replication, LMP2 may act to maintain EBV in a latent state of infection.18 Reversion to type III latency likely occurs at some low frequency since normal individuals maintain lifelong strong cytotoxic T-cell responses against type III latency antigens. In fact, a recent study suggested that type III latency is frequently observed in tonsillar B cells of normal hosts.19 In immunocompromised hosts, the balance between immune clearance and B cell proliferation is disturbed. The lymphoproliferative syndromes seen in these hosts probably represent the in vivo equivalent of LCL transformation.
Primary Infection
For obscure reasons, primary EBV infection is accompanied ∼90% of the time by the appearance of antibodies that react with antigens found on sheep, horse, and beef erythrocytes. Detection of these so-called heterophile antibodies forms the basis for commercial assays such as the Monospot test.20 Heterophile antibodies can be found in 5–10% of the healthy adult population, and in the setting of immune dysregulation by HIV, the rate may be even higher. In children, the only HIV-positive population likely to be EBV-naive, primary EBV infection is heterophile negative in about one-half of cases. When diagnostic uncertainty exists, specific antibodies to EBV proteins can be measured.
Immunoglobulin M (IgM) antibodies to viral capsid antigen (VCA) are present in 90% of acute infections and absent in the general population, thus their presence is essentially diagnostic of primary infection.20,21 By contrast, a fourfold rise in VCA IgG titers can only be demonstrated in 10–20% of cases because titers are generally already high upon initial presentation. Measurement of other antibodies is probably of limited clinical utility. One possible exception is that seroconversion to anti-EBNA antibodies occurs relatively late and can be used to confirm recent EBV infection in a patient previously documented to be anti-VCA positive and anti-EBNA negative.22 Although virus can be cultured from the saliva, this assay is of little clinical use since it is slow, unable to distinguish acute infection from the viral shedding seen in healthy adults, and not generally available. Because primary HIV infection can closely mimic primary EBV infection, measurement of an HIV viral load should always be considered in any person presenting with symptoms suggestive of infectious mononucleosis.23
Oral Hairy Leukoplakia
OHL presents as a corrugated or ‘hairy’ white lesion on the lateral surface of the tongue that is not removed by gentle scraping. It is a nonmalignant lesion caused by unchecked lytic replication of EBV.24,25 OHL is seen in ∼20% of persons with asymptomatic HIV infection and becomes more common with advanced disease.26 Development of OHL is itself associated with a more rapid progression to AIDS and death after controlling for CD4 count.27,28 It is also seen in other immunosuppressed persons, including bone marrow and solid organ transplant recipients.29,30 OHL can very rarely be seen in the absence of immunosuppression, nevertheless, its presence demands that HIV be excluded.31 The diagnosis of OHL is generally based on the typical appearance of the lesions in the appropriate clinical setting (Fig. 51-3). The differential includes oral candidiasis which can be distinguished by its ease of removal from the tongue and/or an empiric trial of antifungal therapy. Biopsy for histology and in situ hybridization or immunofluorescence staining for EBV is rarely necessary, but will confirm the diagnosis. PCR detection of EBV in ‘oral scrapes’ is neither sensitive nor specific for OHL.32
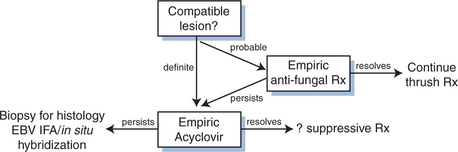
Persons with OHL are frequently asymptomatic and lesions spontaneously resolved in 37% of a cohort followed for 1 year.33 The incidence of OHL has decreased since the advent of HAART and antiretroviral therapy should be sufficient in most cases.34 Further selection of therapy for OHL is based on data from small case series as large placebo-controlled trials are unlikely to ever be completed. An early study by Resnick et al treated six of 13 OHL patients with acyclovir (3.2 g/day for 20 days), and observed regression in five.35 All five recurred with cessation of therapy, and no spontaneous regressions were seen in the seven who refused therapy. Other authors have reported similar results.36–38 Desciclovir (750 mg/day), an analog of acyclovir, produced a similar complete response followed by relapse in eight patients treated by Greenspan et al.39 Foscarnet and ganciclovir which also inhibit EBV replication, have been associated with resolution of OHL in the context of treatment of cytomegalovirus-related disease.40,41 Less impressive results were seen in a study of acyclovir for CMV prophylaxis, 19 of 32 (59%) who had OHL on entry improved on acyclovir versus 13 of 30 (43%) in the placebo group.42 Moreover, there appeared to be no prophylactic benefit: the incidence of new OHL in the placebo group was 16% compared to 14% in the acyclovir group (118 patients in each group).
Topical therapy of OHL has also met with some success. Treatment with 25% podophyllum resin in one study resulted in remissions lasting 2–28 weeks.43 A second observer-blinded study demonstrated marked improvement (relative to the untreated side) within 2 days of a single application.44 Side effects were minimal, and consisted of slight burning or pain and transient dysgusia.
NHL
NHL is an AIDS-defining illness and persons with AIDS have an ∼100-fold increased risk of NHL (see also Chapter 60).45,46 Because the incidence of NHL has not decreased as dramatically as opportunistic infections, it is responsible for a greater proportion of the AIDS attributable morbidity and mortality in the era of HAART.47–51 Risk of NHL is consistently associated with low CD4 count, but increasing age, and male sex have also been implicated.48,49,51 Selective loss of EBV reactive T cells may promote the clonal expansion of EBV-infected B lymphocytes and, in conjunction with other factors, result in lymphoma. Indeed, loss of EBNA1 reactive CD4+ and CD8+ T cells has been documented in AIDS patients with NHL.52 Other factors such as polyclonal B-cell expansion from chronic antigenemia may explain higher rates, particularly of Burkitt lymphoma, seen in less immunocompromised HIV-infected persons.53
HIV infection is associated with two types of EBV-related B-cell lymphomas. The first, diffuse large cell lymphoma (DLCL), bears a striking resemblance to post-transplant lymphoproliferative disease (PTLD). As with PTLD, it occurs in the setting of profound immunosuppression; those with the lowest CD4 counts for the longest time are at greatest risk. Presentation as primary CNS lymphoma is frequent and essentially all CNS lymphomas are EBV positive, whereas about two-thirds of DLCL outside the CNS is EBV positive.54 The frequency of type I verses type II EBV in these tumors roughly parallels the increased prevalence of type II EBV infection seen in the HIV-positive population.55–57 The pattern of EBV latent gene expression in DLCL and PTLD resemble that seen in LCLs.58–60 Both diseases probably represent EBV-driven B-cell proliferation that has escaped immune control.
The second EBV-related B-cell lymphoma seen in HIV infection, Burkitt-like lymphoma, generally occurs early in the course of HIV infection and is EBV positive 30–40% of the time.54,61 Presentation at multiple extranodal sites and aggressive growth are the norm. The tumors contain the typical c-myc translocations of Burkitt lymphomas.62 As with classic Burkitt lymphoma, the precise role of EBV in pathogenesis is not clear. When present, EBV episomes are monoclonal, implying that EBV infection preceded tumor expansion.63 However, latent EBV gene expression in these tumors is restricted to EBNA-1 and the EBERs which are expressed do not have well-established oncogenic properties.
The diagnosis of EBV-related NHL generally rests on the histology and in situ hybridization for EBV gene products of appropriate biopsy material. Amongst solid organ and bone marrow transplant recipients, a consistent correlation between serum EBV viral load and risk of PTLD is observed.64–68 Limited data suggest that this correlation extends to DLCL in HIV-positive patients.69 Not surprisingly many persons without DLCL have detectable amounts of EBV DNA in their serum. Moreover, EBV serum viral load can correlate poorly with clinical response during treatment.70 Further studies are required before a role for serum EBV PCR can be entertained in HIV-infected patients.
The approach to CNS mass lesions in AIDS is presented in Figure 51-1. Because the magnetic resonance imaging (MRI) and computed tomography (CT) appearance of primary CNS lymphoma are not sufficiently specific to exclude infectious etiologies, single photon computed tomography (SPECT) or positron emission tomography (PET) scanning may be desirable.71,72 Unlike the results seen for serum, PCR for EBV DNA in the cerebrospinal fluid (CSF) has high sensitivity and specificity (>90%).73–75 It has been suggested that the combination of PET scanning and EBV PCR may avoid the need for most biopsies.71 However, as primary CNS lymphoma is seen only in the profoundly immunosuppressed, this diagnosis should be confirmed by biopsy in patients with CD4 counts >100/μL.
The prognosis of AIDS-related DLCL had been quite poor, but has improved significantly with the advent of HAART.49,76–78 Treatment of NHL outside the CNS begins with a complete staging workup including: complete blood count (CBC), lactate dehydrogenase (LDH), CT of abdomen, pelvis, and chest, bone marrow biopsy, and lumbar puncture (to exclude concurrent leptomeningeal disease). Systemic chemotherapy is required, even in cases that appear to be localized. A complete discussion of NHL chemotherapy is presented in Chapter 60, but several points are worth noting here. In general, dose-reduced chemotherapy regimens have produced similar response rates and less myelosuppression and infectious complications than standard doses.79,80 Similarly, the anti-CD20 monoclonal antibody rituximab which has established efficacy and safety for treatment of NHL in HIV-negative patients, failed to improve clinical outcome in a phase III trial of AIDS-associated NHL.81–83 Although rituximab administration appeared to improve response rates, this was offset by an increase in infectious complications not seen in earlier phase II trials. It remains possible that select patients, particularly those with the most preserved immune function, may benefit from dose intensified chemotherapy or rituximab administration. Treatment of primary CNS lymphoma remains problematic with reported median survival rates <2 months.84 Radiation therapy can increase median survival to ∼4 months and dramatically improve quality of life. The role of systemic chemotherapy for primary CNS disease remains to be defined.
Perhaps the most controversial aspect of treatment of EBV-associated lymphoma is the role of antiviral therapy. Despite strong evidence that latent EBV infection is central to the pathogenesis of these malignancies, interest in antiviral therapy persists. Several justifications for this interest have been advanced. First, PTLD in pediatric patients frequently occurs in the setting of primary EBV infection. The use of acyclovir (or similar agents) in this setting may limit the extent of spread of the virus to the B-cell compartment. It has also been argued that lytic gene expression, reported in some tumors,85 may allow antivirals to act against some fraction of EBV-positive tumor cells. Several investigators report successful treatment of EBV-associated lymphomas with regimens that include acyclovir, foscarnet, or ganciclovir, though no well-controlled trials exist to support or refute this practice.86–89 A retrospective study by Fong et al, suggested that acyclovir may find a role in the prevention of EBV-associated lymphoma.90 They found patients receiving high, low or intermittent doses, or no acyclovir developed NHL at a rate of 7%, 16%, and 25% respectively. The distribution of CNS lymphoma (3%, 9%, and 5%) did not correlate with acyclovir exposure, and the proportion of non-CNS lymphomas that were EBV positive was not determined. An earlier study found no decrease in NHL associated with acyclovir exposure.91 Such a prophylactic effect, if confirmed by prospective trial, would be surprising in light of earlier data that acyclovir treatment has no effect on number of circulating EBV-positive B-cells.92
One antiviral drug that has shown activity against EBV during latent infection is hydroxyurea. In vitro this drug can eliminate EBV genomes from tissue culture cell lines. Slobod et al reported a limited response to hydroxyurea in two patients with primary CNS lymphoma.93
Other EBV-Associated Diseases
Lymphoid interstitial pneumonitis is characterized by diffuse interstitial pulmonary infiltrates. It occurs primarily in children infected with HIV, but can also be seen in adults. EBV proteins and DNA have been detected in biopsy specimens from affected children.94
EBV genomes have also been found in leiomyosarcoma biopsies from children with AIDS.95 The authors were able to demonstrate that these tumors expressed the EBV receptor (CD21) and did not find any evidence that EBV was associated with smooth muscle tumors of HIV-negative patients.
Two other EBV-associated diseases deserve mention. Nasopharyngeal carcinoma (NPC), a disease endemic in southern China and among the Inuit, is strongly associated with EBV infection.2 There does not appear to be an appreciably increased incidence of NPC in the HIV-positive population. Hodgkin disease, particularly the mixed cellularity and lymphocyte depleted forms, has also been linked to EBV infection. There is probably a modest increase in risk for HIV-positive persons in whom the disease tends to follow a more aggressive course.96
KSHV
Epidemiology
Although advances in the molecular biology of KSHV have outpaced our understanding of its natural history, epidemiologic studies were critical to establishing an infectious etiology for KS. A transmissible agent had long been suspected and intensive investigation had even uncovered ultrastructural evidence for a herpesvirus in KS tissue.97 However, repeated efforts failed to isolate a pathogen. This changed in 1994 when Chang, Moore, and colleagues, using a novel technique called representational difference analysis, succeeded in isolating two DNA sequences from KS tumors that ultimately proved to be from KSHV.98 They later stated that epidemiologic evidence was a critical determinant of their decision to continue to search for a ‘KS agent’.99 Particularly compelling were surveillance data from the CDC which established that men who have sex with men (MSM) were 20 times more likely to present with KS than similarly immunodeficient hemophiliac men.100,101 Remarkably, much of what we know about the natural history of KSHV infection in HIV-positive persons, could be deduced from study of the epidemiology of KS. Infection with the KS agent is relatively uncommon in the general population, and it is transmitted by sexual or parasexual practices common amongst MSM, but inefficiently transmitted by blood or blood products.
The identification of a specific pathogen enabled investigators to prove the same virus was associated with all forms of KS: classic, endemic, AIDS, and transplant related.98,102 The diversity of assays used to measure KSHV antibodies has made estimating KSHV seroprevalence anything but straightforward. For example, assays that measure antibodies to latent KSHV antigens yield prevalence rates consistently lower than assays that measure antibodies to lytic antigens. Nevertheless, it can be safely stated that KSHV infection is not ubiquitous and displays distinct geographic variation. The KSHV seropositivity in the general population of the US and Western Europe is in the range of 1–5%.103–105 In certain regions of Mediterranean countries where classic KS is found this increases to 10–20%,105–107 and may be as high as 30–80% in parts of sub-Saharan Africa.108–114 In Western countries, KSHV seroprevalence among HIV-negative MSM is ∼11–20% and rises to the 30–54% range in HIV-positive MSM, but amongst other HIV risk groups is comparable to that seen in the general population.104,115–117 One retrospective study found that a cohort of HIV-positive MSM seroconverted to KSHV at a median of 33 months prior to the development of KS.103
Although a receptor for KSHV has recently been identified, the precise mode of transmission of KSHV remains unclear.118 The marked geographic variation in seroprevalence indicates that transmission is inefficient relative to herpesviruses such as EBV. In endemic countries, acquisition in childhood appears to be common, indicating that the mode of transmission may be different than in the MSM population. The observed clustering in families suggests transmission between siblings or from mother to child may occur.119,120 Although reported, neither vertical transmission nor transmission through breastfeeding appears to be the major mode of transmission because seroconversion occurs predominately after the age of two.121,122 Studies of MSM cohorts have suggested HIV positivity, increasing number of sexual partners, a history of a partner with KS, orogenital sex, and even use of inhaled amyl nitrate capsules were independent risk factors for KSHV seroconversion.116,123–127 In heterosexual populations, birth in Africa appears to be the strongest risk factor.124 Some studies have observed elevated rates of KSHV positivity at STD clinics or amongst female sex workers, though other studies did not find evidence for heterosexual transmission.104,105,124,128,129 KSHV DNA was found in the semen of HIV-infected persons, but was not in samples from healthy donors.130 However, viral loads appear to be about two logs higher in saliva than in semen, breast milk, or blood, and infectious virions have been recovered from saliva specimens.131,132 These findings are most consistent with salivary transmission of KSHV.



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


