Epigenetic contributions to human cancer
James G. Herman, MD  Stephen B. Baylin, MD
Stephen B. Baylin, MD
Overview
Over the past 20 years, an exciting advance in our understanding of the mechanisms underlying cancer development has been our growing appreciation that these diseases are not driven solely by genetic changes, but also by epigenetic changes.1–4 Strictly speaking, the term epigenetic refers to heritable changes in gene expression, in dividing somatic cells, which are mediated by alterations other than changes in the primary base sequence of DNA.5–7 This definition encompasses two critical translational characteristics concerning epigenetic alterations in cancer and their clinical importance. First, the coding and noncoding genes affected by epigenetic changes in cancer remain wild type for DNA sequence rather than harboring irreversible mutations. Second, and closely related, the changes are, then, potentially reversible if normal gene expression can be restored such that the wild-type gene function can emerge.1, 2, 4, 7–10
This recognition of epigenetic changes, which are fundamental to cancer initiation and progression, is occurring in the midst of a dynamic explosion of knowledge as to how the human genome is normally controlled, via chromatin packaging of DNA, to regulate gene expression in different tissues and during development.5–7, 11 For example, epigenetic processes play a fundamental role during normal embryonic development and adult-cell renewal. As such, these processes control the emergence of different cellular phenotypes, all with the exact underlying DNA sequence, which occur during development and differentiation. While this knowledge of gene regulation continues to grow rapidly, our understanding of the complete spectrum of epigenetic changes that are key to cancer development is expanding rapidly but could even still, relatively, be in its early stages. However, there is much that has already been learned, and this knowledge in the understanding of basic cancer biology and carcinogenesis already has palpable translational implications, which are discussed in this chapter.
Mechanisms involved in epigenetic regulation of gene expression
Formation of chromatin
While all of the fundamental information for gene expression lies in the primary base sequence of DNA, which can be viewed as a “hard drive” for storage of this essential coding, the patterns of gene expression in cells are determined by how this DNA is modified following synthesis and replication and how it is packaged into the nucleus by the proteins, or chromatin, by which it is arranged.5–7, 11 The latter processes of packaging might, then, be viewed as the “software,” which provides the readout of the hard-drive information contained in the DNA sequence. The primary role of the DNA modification and chromatin packaging is required to balance the genome such that the majority of DNA is encompassed in a silent or low-transcription state to guard against unwanted expression of repeat sequences, potential transposable elements, and viral insertions accrued over evolution.5, 6, 12 The first element of DNA packaging is its interactions with proteins forming chromatin. The fundamental scaffolding proteins for chromatin are the histone proteins, and their assembly with DNA, in turn, forms the key element, or nucleosomes (Figure 1), which are essential for arranging DNA in the nucleus.13, 14
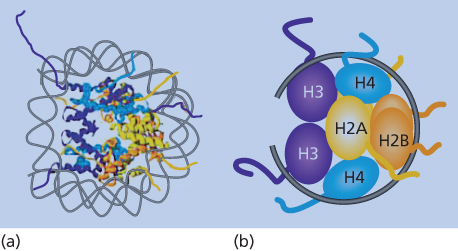
Figure 1 Nucleosome structure. (a) Model of the double helix wound around the protein structure of the constituent histones as outlined in the text. (b) The schematic representation shows the organization of the H3/H4 tetramer on the DNA, followed by two sets of H2A/H2B dimers forming the histone core with the DNA (black line) wrapped around. The amino-terminal histone tails are shown extruding from the nucleosome core of the eight histone proteins.
Nucleosomes consist of ∼146 bp of DNA wrapped twice around an octamer of the core histone proteins, H2A, H2B, H3, and H4.13, 14 For functional mediation of gene expression profiles from DNA, nucleosomes must not only be properly distributed linearly along DNA, but must also be arranged into higher-order, multinucleosome structures13, 14 (Figure 2). These dynamics are mediated by chromatin-remodeling proteins, and the more widely and irregularly spaced the nucleosomes are in their linear placement along DNA, and the less compacted the higher-order structures, the more “open” the chromatin is and the more available the DNA is for active-gene transcription (Figure 2). This is often the case in areas in and around active-gene promoters. Conversely, the more regular and evenly the nucleosomes are spaced, and the more compacted their higher-order structures (Figure 2), the more “closed” is the chromatin and the more repressive for gene transcription. This latter configuration dominates most of the human genome, as noted earlier, to prevent unwanted gene expression and to facilitate chromosome structure.
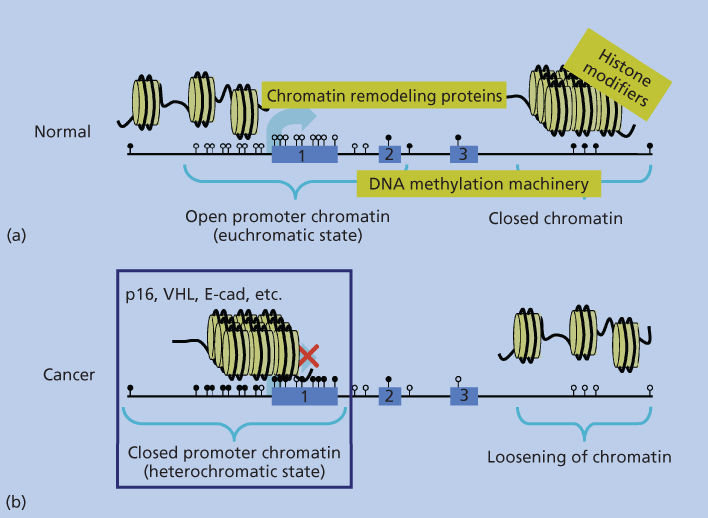
Figure 2 Depiction of the packaging arrangement of human DNA (straight black line). Panel (a) shows, for normal cells, a more linear arrangement of nucleosomes (yellow circles with DNA wrapped around as outlined in Figure 1) depicting the “open” arrangement of chromatin around most gene promoters containing CpG islands (unmethylated CpG sites shown as white lollipops), which are being actively transcribed or can be induced to transcribe (large light-blue arrow). Exons 1–3 of the model gene are depicted by the numbered light-blue boxes. Regions of DNA within the body of the gene and extending beyond the third exon are depicted as being in the closed, transcriptionally repressed, conformation typical of the majority of the normal human genome. This closed structure is represented by the more packed, three-dimensional organization of the nucleosomes, and the CpG sites in the closed regions are depicted as being methylated (black lollipops). The text in yellow squares depicts the chromatin remodeling protein complexes, histone-modifying enzymes, and enzymes performing DNA methylation (DNA-methylation machinery), which are responsible for DNA packaging as discussed in the text. Panel (b) depicts the altered chromatin patterns present in a typical cancer cell with a switch in positions of the normal closed and open chromatin regions in the genome. Many closed regions now have an open chromatin with loss of DNA methylation, while a large group of gene promoters have assumed a closed chromatin with abnormal, CpG island DNA methylation and repressed transcription (large red X over the transcription start site depicted by the light-blue arrow.
Integrally involved with this dynamic chromatin structure is a dependency of nucleosome function on states of chromatin, which are determined by differing ratios of active and repressive histone “modifications.”6, 7, 11, 14, 15
These consist of additions or modifications to key amino acids, primarily located in the tails of the histones that stick out from the nucelosome assembly, in the form of lysine acetylation, lysine and arginine methylation, serine and threonine phosphorylation, glutamic acid ADP-ribosylation, and lysine ubiquitination and sumoylation.6, 7, 11, 14, 15
The balance of these marks form what was initially termed a “histone code,” now realized to be more complex than envisioned, that participates, along with nucleosome positioning, in packaging the genome such that both constitutive and cell type-dependent chromatin patterns of open and/or closed configurations are maintained from cell division to cell division and ensuring that these patterns remain stable in nondividing cells.6, 7, 11 It is in this way that cells maintain a “memory” for patterns of gene expression and chromosome structure that facilitate normal patterns of development and the maintenance of mature-cell renewal and differentiation states.14, 15
The best characterized of the histone modifications, mentioned earlier, are lysine methylation and acetylation, which are most closely associated with either high or low states of gene transcription. These marks in turn are established by families of enzymes consisting of histone methyltransferases (HMTs) that catalyze the methylation, histone demethylases that remove these methyl additions, and histone acetylases or (HATs) and histone deactylases (HDACs), which place and remove the acetyl groups, respectively.7, 11, 15–17 For these dynamic events, examples of key methylation marks associated with gene states are methylation of lysine 4 of histone 3 (H3K4me3), which is associated with open chromatin typical of active transcription, and methylation of lysine 9 or lysine 27 (H3K9me3, H3K27me3), which is characteristic of repressed-gene expression.6, 15 Adding to the information contained in this histone code is the fact that lysine methylation may be present in either monomeric, dimeric, or trimeric, states.6, 7, 11, 15 There is also a regulated balance for histone acetylation in which enzymatic activities of the HATs and HDACs determine the states of histone–lysine acetylation that are typical of open, transcriptionally active chromatin, and deacetylation, more associated with closed chromatin and repressed transcription.7, 11, 15 Key examples of such active marks at gene promoters are H3K9acetyl and H4K16acetyl.7, 11, 15
DNA methylation
While not present in all multicellular organisms, humans, other mammals, and other higher organisms add an additional layer of epigenetic regulation. Working in close concert with chromatin states to package the human genome is a key modification consisting of methylation added directly to postreplicated DNA.7, 10, 17, 18 This step consists of attachment of a methyl moiety to the C5 position of the base, cytosine, only when it is located preceding a guanine or in a “CpG” dinucleotides context. The methyl group is transferred from S-adenosyl methionine to DNA through catalysis by a family of DNA methyltransferase (DNMT) enzymes.17–19
The role of this DNA methylation19 is closely tied to the distribution of the CpG dinucleotide in human and other genomes. This is a nonrandom and uneven distribution in which there has been a global and progressive depletion of CpGs over evolution because the deamination of methylated CpGs results in changing the cytosines (Cs) to thymidines (Ts).7, 17, 18 Failure to repair these thymidines then results in transition of the Cs to Ts. However, there remains interspersed conservation of nondepleted, CpG-rich stretches (∼0.4 to several thousand kb) or the so-called “CpG islands,” which are particularly important to the DNA-methylation patterns.17, 18 These islands, especially when found in the 5′ end of about 50–60% of human genes,17, 18 remain non-DNA methylated, while the majority of the CpG′ sites in the remainder of DNA are methylated4, 7, 17, 18 (Figure 2).
This pattern of DNA methylation, depicted in Figure 2, works in tight concert with the nucleosome positioning and histone modifications previously discussed to determine the epigenetic regulation of the genome. Thus, methylated DNA associates with, and helps maintain in a tight heritable state, the relatively transcriptionally inert status of the majority of the genome, which is most apparent in the closed chromatin or “heterochromatic regions” concentrated in pericentromeric parts of chromosomes. In contrast, the non-DNA-methylated CpG islands associated with gene-start sites appear to reflect and facilitate a transcriptional ready and/or active transcription state.4, 7, 17 This tight interaction between histone modifications and DNA methylation is reflected in the fact that transcriptional repression is associated with DNA methylation, which serves to help maintain the repression of many closed chromatin states. Thus, deacetylated histone lysines, such as for H3K9, and repressive methylation marks, such as H3K9me3, associate with methylated DNA.17–19 In turn, such marks, and particularly H3K9me, appear important for targeting of DNA methylation.17–19
It is important to note that an important exception to the above role of DNA methylation in repression of gene transcription has been recently noted. Thus, one common site of 5mC is in the body of active and not repressed genes. This methylation appears to work with the mark of H3K36 me3 to allow transcriptional elongation and enhanced gene expression.7, 10, 20, 21
There have been recent and most important advances in our understanding of how DNA-methylation patterns are established and maintained for inserting and maintaining DNA methylation in the epigenome. It has long been recognized that three biologically active enzymes, DNA methyltransferases (DNMTs), are responsible for establishing and maintaining sites of DNA methylation.19 In this regard, DNMT 1 is predominantly a maintenance DNMT, which is responsible for preserving patterns of established DNA methylation during DNA replication. DNMT3A and DNMT3B are predominantly de novo DNMTs, which can establish new sites of DNA methylation. However, much data suggests a more complex scenario in which there can be cooperativity between the three DNMTs and interacting proteins such that they can, in some instances, replace one another in function when stress situations require this.22 For example, DNMT3A and DNMT3B may function to repair errors made by DNMT1 during DNA synthesis.14
While the above scenarios for establishing DNA-methylation patterns have been appreciated, it was long thought that removal of this modification is a passive event accomplished only by failure to replace methylation sites during DNA replication. However, an exciting new era has emerged in which we appreciate that, as for histone modifications, there are distinct active steps for erasing DNA methylation. A family of enzymes, termed TET (ten-eleven translocations) family proteins, can, through oxidative steps, convert 5-methyl cytosine (5MT) to 5-hydroxylmethyl cytosine (5hMT), which, through subsequent DNA repair steps, convert 5MT back to cytosine.19, 23, 24 These conversions are important during normal development and adult-cell functions, and later we outline their importance for cancer.25–27
Altered DNA methylation and chromatin in cancer or the “cancer epigenome”
Loss of DNA methylation
Virtually all cancer types harbor what appears to be a marked shift away from the normal epigenetic patterns, described in the previous sections, for normal cells4, 7, 8 (Figure 2). This has been best studied to date for DNA methylation, where there are at least two major changes that are now well appreciated. First, there are global losses of this modification from the widespread regions of the genome, which harbor DNA methylation in normal cells1, 2, 7, 8, 10 (Figure 2). Indeed, this was the first chromatin abnormality well cataloged for cancer28 although much remains to be learned about the ramifications of this change. As these are generally areas of closed chromatin where the DNA methylation helps to maintain transcriptionally repressed DNA, such losses could associate with abnormal transcription.1–4 Indeed, a number of genes with oncogenic potential, and which normally have low expression in normal cells, have now been reported to be upregulated in association with cancer-specific decreases in promoter DNA methylation.1 Moreover, pericentromeric regions are a target for DNA-methylation losses in multiple cancer types and this may play a role in the genesis of chromosomal instability in neoplasia.1–4, 29 Most recently, losses of DNA methylation from gene body regions have also been observed in tumors. This can have an opposite effect on gene expression to those above as methylation in these regions, as discussed earlier, actually facilitates gene expression through enhancing transcriptional elongation.7, 20, 21
Gene promoter DNA hypermethylation
The best-studied chromatin change associated with epigenetic abnormalities in cancer, and the one with the most-recognized ramifications, to date, entails localized increases in DNA methylation in gene promoter CpG islands, which are protected from this change, as discussed earlier, in normal cells (Figure 2).1–4, 7, 8 This change is associated with tight transcriptional repression of genes and can, thus, serve in a process that provides an alternative to gene mutations for loss of function of a number of well-characterized tumor-suppressor genes.1–4, 7, 8 In addition to these classic-suppressor genes, data derived from random screens of cancer-cell DNA for DNA-hypermethylated genes, such as in the Cancer Genome Atlas Project (TCGA), indicate that hundreds of such genes appear to exist, for multiple cancer types analyzed, in a given patient’s tumor.1–4, 7, 8, 30–33 While all of these gene changes may not be pivotal for driving the initiation or progression of the particular cancers that harbor them, many, including the classic-suppressor genes, do encode for genes for which loss of function would be important for tumor development.3, 4, 7, 8 Many genes, which are seldom or never mutated in cancers, may still have important roles in tumorigenesis, as they undergo promoter DNA hypermethylation and silencing during tumor progression.4, 7, 8 In this regard, virtually every critical pathway known to play a role in tumorigenesis is now known to be involved with genes bearing cancer-specific DNA hypermethylation in one or more tumor types3, 4, 7, 8 (Table 1). Finally, many of the genes involved exhibit the DNA-methylation change in preinvasive lesions, which have the potential for malignant progression, as demonstrated by “benign” colon polyps.1, 2, 4, 7, 8, 34 In these preinvasive colon lesions, multiple hypermethylated genes are already present and encode for key genes that, when inactivated, would deregulate key pathways, such as the Wnt pathway, which are well known to drive the initiation and progression of all colon cancers.1, 2, 4, 7, 8, 34 These findings have led to the hypothesis that epigenetic changes may be in many cases important for tumor initiation and for the appearance of abnormally expanding cells, which arise in cancer-risk states, such as chronic inflammation.4, 7, 34–36 In addition to the above findings, it is now increasingly appreciated that differential DNA increases may affect key genomic regions other than just gene proximal promoter regions.3, 4, 7, 14, 19, 37, 38 Regions just distal to promoter CpG islands called “shores” can be differentially methylated between normal and cancer tissues.37, 38
Table 1 Examples of pathways altered by gene-promoter hypermethylation and gene silencing in cancer
| Pathway | Genes 3–5 |
| Wnt pathway | APC, SFRP family, SOX17 |
| Altered cell-cycle control | Rb, p16, p15, p14, p73 |
| Repair of DNA damage | MLH1, O6-MGMT, GST-Pi, BRCA1 |
| Apoptosis | DAP kinase, caspase 8, TMS-1 |
| Tumor-cell invasion, angiogenesis | THBS1, E-cadherin, VHL, APC, LKB1, TIMP3, |
| Tumor architecture | Growth-factor response ER, RAR-beta, SOCS-1* |
The above shore regions are also differentially methylated between normal tissues, so the meaning for cancer per se, and especially as reflective of associated gene expression changes, is less clear than for proximal promoter, CpG island regions. Similarly, there is a building body of data for increased methylation in gene-enhancer regions, or DNA-regulatory sequences, which through binding of key proteins and histone modifications are master regulators of gene expression. These sequences can lie at variable, and often large, distances away from gene proximal promoters.39–41 De novo DNA-methylation changes at enhancers may also contribute key gene expression abnormalities in cancer and especially for increasing cancer-risk states.42, 43 While the theme of differential enhancer DNA methylation in cancer is under more and more investigation, the role of these changes, and their balance with gene promoter CpG island hypermethylation, is still being clarified.
Long-range changes in DNA methylation and chromatin in cancer
A recent exciting advance in studying both normal and cancer epigenomes, stemming from in-depth analyses of CpG methylation and chromatin throughout the genome, is that there are nonrandom regional configurations that can be altered between cancer and normal cells. These occupy megabase regions in many chromosomes (100 kb to 10 Mb).3, 4, 7, 8, 44–46 These are largely CpG poor regions, and the constituent CpGs are heavily methylated but to degrees that vary significantly throughout different normal tissues leading to the term partially methylated domains.3, 4, 7, 8, 44–46 Cancers characteristically have losses of normal DNA methylation throughout these areas, creating the so-called megabase islands or canyons of hypomethylated domains as particularly defined to date in colon and other cancers.3, 4, 7, 8, 44–46 Within these blocks of sequence, cancers can establish long-range appearance of repressive histone modifications, such as for H3K9 methylation as well.3, 4, 7, 8, 44–46 In some regions, there may be long-range areas of nonrepressive or more open chromatin.45, 47–50 Intriguingly, much smaller-embedded genes within these large domains, which harbor promoter CpG islands, can have the de novo gains in DNA methylation discussed in detail earlier.3, 4, 7, 8, 45 Thus, in essence, hypomethylated canyons in cancer often exhibit many of the cancer-associated losses and more focal CpG island gains of DNA methylation in cancer.3, 4, 45 Moreover, these large domains may harbor a much higher than expected percentage of genes with a history of embryonic and adult stem cell chromatin regulation that are particularly vulnerable to abnormal, promoter CpG island DNA hypermethylation.3, 4, 7, 8, 35, 45, 51, 52
The increasing connections between genetic and epigenetic alterations in cancer
One of the most intriguing themes to emerge over the past several years is the recognition that, in virtually all tumor types, one of the most frequent type of mutations is in genes encoding for proteins that establish and maintain the epigenome.3, 4, 7, 53, 54 While the exact implications of these remain to be defined, there are now several key links between these genetic alterations and either tumorigenesis, DNA-methylation abnormalities, or chromatin changes. One important example concerns the relationships between mutations in IDH 1 and 2 genes, and also in the TET genes, and DNA hypermethylation. As now recognized in low-grade gliomas in younger patients and in hematologic cancers,55–57 these mutations associate with altered DNA and histone-demethylation pathways. These can include changes in histone-methylation levels and a pattern of increased frequency of promoter region CpG island DNA hypermethylation, which resembles what was initially characterized, in a subset of colon cancers, as the CpG island methylator phenotype or CIMP.56–59 Much is coming to be understood about why the IDH mutations, especially, produce chromatin and DNA-methylation changes. The mutated genes lead to the massive accumulation of 2-hydroxy-glutarate formed from, and leading to depletion of, ketoglutarate.60–62 The latter is a key cofactor for multiple enzymes that help regulate and maintain key chromatin marks and the previously mentioned TET proteins that can protect against abnormal DNA methylation.63, 64 These changes are thought to incite, over time, a molecular progression from increases in repressive-histone marks and subsequently the above DNA hypermethylation.60–62 Recent mouse studies indicate that IDH mutations are drivers for early tumor-progression events65 by blocking normal differentiation of normal stem/progenitor cells and thus facilitating abnormal self-renewal and diminished lineage commitment and differentiation.57, 60, 66 Importantly, drugs directly targeting the IDH mutations have been developed and are now undergoing trials in AML and brain tumors.
Another fascinating set of mutations has been delineated in a pediatric subtype of brain tumor in a key repressive, histone mark itself. Thus, mutations in H3K27, present in only one allele of the multiple copies of histone H3, block the activity of the enzyme, EZH2, which catalyzes H3K27 methylation. Accordingly, the tumors have a marked loss of H3K27me3,3, 67, 68 and this is postulated to induce abnormal activation of genes to drive initiation and/or progression of the tumors.
Clinical implications of altered DNA methylation in cancer
Although the full impact of understanding chromatin alterations in cancer is still to emerge, important progress is being made in at least two broad categories of translational application. First is the use of DNA-hypermethylated gene promoter sequences as tumor biomarkers, and second is the use of epigenetic therapies, which may be efficacious because they target reversal of abnormal gene silencing.1, 2, 9, 69–71
Cancer DNA methylation biomarkers
There are some simple advantages that the detection of DNA-methylation changes in cancer allows over other molecular approaches. First, the overall frequencies of methylation changes appear to be greater in many instances than mutational changes.3, 7, 30, 69 This appears to be true for all major solid and liquid cancer types examined thus far, although the specific genes altered are different among tumor types. A second advantage of the detection of DNA-methylation changes associated with gene silencing is that this change is confined to the promoter region for many genes, making this alteration easily targeted for assays that can potentially serve as molecular markers for cancer. The large numbers of these genes allow for construction of gene panels for which DNA-hypermethylation markers essentially cover cancer genomes providing for high chance of marker detection in any given patient.72 The fact that the abnormality arises for many genes in early, preinvasive stages of cancer and for others later in progression allows for potential uses of such markers in cancer-risk assessment, early diagnosis, molecular restaging of tumors, and prediction of tumor behavior. Thus, the high frequency, early occurrence of such changes, and the ability to assay methylation of each gene with a single assay, facilitate the use of such approaches for the early detection of cancer. Examples of each of the marker possibilities mentioned earlier include those listed in Table 2. Thus, for cancer-risk assessment, detection of DNA-hypermethylated gene sequences in sputum DNA now holds considerable promise for predicting which individuals at high risk for developing lung cancer will actually manifest this disease within a defined period after initiation of marker monitoring.2, 73 Detection of colon cancer has been explored by detection of altered DNA methylation in either the blood or stool and these are emerging as especially useful when added to mutation detection.74–79 Detection of such genes in urine can potentially stratify the risk for, or provide for early diagnosis of, bladder cancer80 and in prostate biopsies for improvement in the diagnosis of prostate cancer.81, 82 Several of the tests, for stool, and prostate biopsies are now available to clinicians via CLIA-approved tests.
Table 2 Examples of using gene-promoter DNA-hypermethylated sequences as cancer biomarkers: nearing clinical use
Early diagnosis and/or detection of high-risk states:
|
Prognosis:
|
Prediction:
|
A second area in which the changes of DNA methylation may be used in the management of cancer follows the observation that these genes are part of key pathways in the development of cancer. This allows the use of DNA methylation in these genes to potentially be used as prognostic or predictive biomarkers (Table 2). In general, the presence of DNA methylation for most genes has been associated with adverse outcome, thus providing a prognostic biomarker. This is consistent with the role that silencing of key tumor-suppressor genes can play in altering key signaling pathways. Thus, tumors with alterations of single genes, or multiple genes, are more aggressive or molecularly advanced, and thus lead to worse outcomes.83, 84 Recently, detection of a small panel of DNA-hypermethylated genes simultaneously in tumor and mediastinal lymph node DNA provides an extremely promising approach to restaging of stage 1 lung cancer patients to stage 3, thus predicting which of these individuals has a high risk of rapid recurrence.85
Another useful tool for the management of patients by medical oncologists is the possibility that DNA methylation in specific genes may affect the cancer-cell sensitivity to different therapies. For example, silencing of a DNA repair gene may increase likelihood of tumor response to DNA-damaging agents that would be repaired by this protein. The most clear example of this has been demonstrated for patients with the virulent brain tumor, glioblastoma, where detection of tumor DNA hypermethylation for the DNA damage-repair gene, O6MGMT, predicts for higher likelihood of response and post-treatment period of freedom from disease recurrence, for patients treated with the DNA-alkylating agent, Temazolamide.86–88 Other studies have suggested that sensitivity to cisplatin may be mediated by silencing of FANCF in ovarian cancer89, 90 and sensitivity of colon cancer to topoisomerase inhibitors may be mediated by silencing of the Werner syndrome gene (WRN).91 Recently, the promoter methylation of SMAD1 has been reported to predict resistance to doxorubicin, for patients with diffuse large B-cell lymphoma (DBCL).92 Low doses of DNMT inhibitors reverse the repression of this gene, and this was found to relieve the chemoresistance.92 These preclinical findings are being tested in a phase I clinical trial with early findings suggesting that the DNMT inhibitor used can prime for better chemotherapy responses.92 Before clinical use, all of these studies need to be confirmed in additional populations and ideally in prospectively evaluated patients. However, such developments might greatly aid in optimizing the therapeutic choices offered to patients, who only have one opportunity for first-line therapy. An ever-increasing number of larger and larger clinical trials seek to validate the marker approaches and push their incorporation into standard oncology practice. Finally, other DNA-methylation changes, such as losses of normal DNA methylation in tumor DNA, and changes in chromatin marks for active or repressed-gene function, will almost certainly appear as promising molecular cancer markers.
“Epigenetic” cancer therapy
This type of therapy refers to cancer treatments based on the concept of reverting abnormal gene expression patterns toward normal as a means of cancer treatment. To date, two classes of drugs, which may achieve this goal, those which can experimentally induce DNA demethylation (5-aza-cytidine, azacytidine and 5-aza-2′-deoxycytidine, or decitabine) and those which inhibit histone deactylases (SAHA), have been approved by the FDA for treatment of the preleukemic state, myelodysplasia (MDS), and cutaneous T-cell lymphoma, respectively. Both azacitidine93 and decitabine7, 94–96 have shown clinical benefit, and most recently, in a randomized phase III trial, azacitidine has shown a survival benefit.97 Although, in both instances, these drugs achieve impressive response rates, and durable responses, in previously refractory diseases, it must be cautioned that their exact mode of efficacy remains to be proven—that is, they may not work clinically, solely, or partially—through the epigenetic effects, they mediate experimentally.70, 95, 98, 99 Thus, use of these drugs in other hematologic malignancies is the focus of many ongoing trials, and potentially promising results are being seen especially for the DNA-demethylating agents in acute myelogenous leukemias (AML) and chronic myelogenous leukemias (CML).94, 100 In addition, there is experimental evidence for synergy of DNA demethylating, and histone deacetylase inhibition, for re-expression of DNA hypermethylated cancer genes.101 Thus, multiple trials are exploring the clinical potential of using combination therapies with these agents. Most of these trials are joined with studies trying to examine whether DNA demethylation and re-expression of abnormally silenced genes corresponds with, or predicts, therapeutic response. Probably, over the next few years, all of these clinical studies will establish the true position of these drugs and their mechanism of clinical efficacy, in the clinical arena.
The use of epigenetic therapy in solid tumors, while less well explored, has been introduced in the past102, 103 and is now becoming a major focus with some promising and important results.70, 71 The universal presence of the therapeutic targets, as discussed in multiple sections of this chapter, makes the potential for such approaches as compelling in these cancers as in the hematologic malignancies. Recent preclinical studies have investigated how low doses of DNMT inhibitors can affect solid tumor cells and suggest that nanomolar doses of both Vidaza and Dacogen can yield antitumor responses, which may reflect the ability of these drugs to “reprogram” cancer cells with little off-target effects.104 Using such low-dose approaches, plus the availability of new DNA-demethylating agents, such as SGI-110, which acts as a prodrug for DAC,105, 106 clinical trials are increasing. These include combining these drugs with other epigenetic therapy agents such as histone deactylase inhibitors (HDACis). For example, such a combination, in recent trials in 65 patients with advanced, pretreated, non-small-cell lung cancer resulted in robust, durable responses in a small subset of patients.107 In addition, results in these same trials suggest that the epigenetic therapy could prime a larger group of patients for better responses to subsequent therapies,107 including standard chemotherapies and exciting new forms of immunotherapy.108, 109 With respect to the immunotherapy, preclinical work suggests that the DNA-methylation inhibitors can, in lung cancer cells and other solid tumor types, induce complex immune cell attraction pathways involving hundreds of genes.110 The potential for the above sensitization of NSCLC patients to chemotherapy and immunotherapy is now undergoing testing in larger in larger trials. In addition to these studies in NSCLC, others are finding that DNA-methylation inhibitors can prime patients with advanced serous ovarian cancer to subsequent chemotherapy.111
It is certain that, over the next few years, many trials will appear for epigenetic-therapy approaches in solid tumors and, as in the leukemias, these will include studies to elucidate the precise mechanisms underlying clinical efficacy. All of these investigations, plus those in the hematologic malignancies, will not only guide the use of currently existing agents, but foster the development of newer and possibly more potent and specific drugs, as well. As we learn more about what precisely mediates abnormal DNA methylation and the components of chromatin that collaborate to initiate and maintain such changes, new molecular targets will almost surely emerge. Combination therapies exploiting these targets will likely follow. In this regard, small molecule inhibitors for several key steps in chromatin assembly have been developed and several of these are already in clinical trials.4, 70, 71, 112–116
Summary
Work over the past decade, especially for DNA-methylation changes, has amply established that, from initiation through progression to advanced stages, cancer is a disease of epigenetic as well as genetic alterations. These studies, closely entwined with an explosion of basic knowledge about how chromatin constituents package the human genome to regulate gene-expression patterns, are providing a rich substrate for new cancer biomarker and therapy strategies. There is enormous potential for these strategies to enter usage in the cancer clinical arena over the next decade.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


