Chapter 23 Enfuvirtide
Prior to the accelerated FDA approval in 2003 of the membrane fusion inhibitor, enfuvirtide (ENF), all available antiretroviral agents were directed against one of two HIV-specific enzymes, reverse transcriptase (RT) or protease. Thus, ENF represented the first clinical agent from a novel therapeutic class in nearly eight years. The advent of an entirely distinct therapeutic class, the viral entry inhibitors, suggested the possibility of avoiding cross-resistance and shared toxicities with previous drugs, and therefore improved prospects for individuals with extensive prior treatment histories. ENF also represented the most complex synthetic peptide ever developed for large scale chemical manufacturing and the first antiretroviral agent licensed solely for administration by the parenteral route, and therefore introduced new challenges in regard to mass production, convenience and adherence, adverse event profiles and the costs of HIV care.
MECHANISM OF ACTION AND STRUCTURE
The process of HIV entry into target cells is complex, involving multiple steps; other viral entry inhibitor candidates in development are discussed in detail elsewhere in this book (Chapters 24 and 27). Briefly summarized, HIV entry into target cells begins with binding of the viral surface glycoprotein (gp120) to the CD4 molecule expressed on host target cells, particularly T helper lymphocytes. The initial interaction between the HIV-1 gp120 and CD4 triggers a conformational change in gp120 to expose a “co-receptor” binding site, with an affinity for chemokines that bind either the CKR5 or CXCR4 class of host receptors.1–3 Following attachment of gp120 sites at both the CD4 and chemokine receptor sites, the viral transmembrane glycoprotein (gp41) undergoes a conformational change that triggers fusion of the viral and cellular membranes. The fusion step allows viral contents to be taken up by the target cell, which ultimately leads to integration into the host nucleus and production of new viral progeny (see Fig. 23-1).
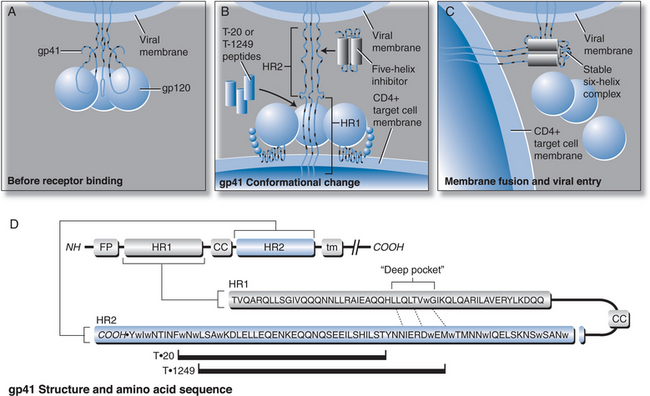
Early studies of the gp41 molecular sequence revealed “heptad repeat” sequences in two regions that give the protein periodic hydrophobicity. These consensus motifs (“leucine zippers”) were predictive of an alpha-helical structure within the gp41.4,5 Synthetic peptides, originally called DP107 and DP178, corresponding to these heptad repeat sequences were found to inhibit HIV infection in vitro.6,7 A series of experiments over the next several years suggested these regions of gp41 form a helical bundle or “coiled coil” structure that is critical for membrane fusion to occur. Mutations in these leucine zipper regions of gp41 were found to disrupt the “coiled coil” structure and thereby interfere with membrane fusion and infectivity. It was proposed that synthetic peptides corresponding to these alpha-helical regions had antiretroviral activity, mediated by disruption of the tertiary protein structure of the protein.8–12
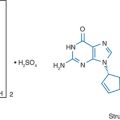
A model of gp41 mediated membrane fusion analogous to the “spring-loaded” mechanism described for influenza virus was proposed.13 When influenza virus first binds to a target cell, hemagglutinin goes through a conformational change, extending from a loop structure to an extended “coiled coil”. This allows a “fusion peptide” to shift into a favorable position so that membrane fusion can occur. The corresponding model proposed for HIV gp41 (Fig. 23-1) is that the fusion peptide is in an unexposed position when gp41 is in its native, “nonfusogenic” state. After gp120 binds to a target cell, gp41 changes conformation, unfolding by a molecular hinge mechanism. The fusion peptide is then extended away from the virus surface and therefore better able to insert into the cell membrane. If the gp41 then returns to its native, folded (“hairpin”) conformation, the result would be to pull the viral and cell membranes into close proximity in order for fusion and viral entry to occur. Based on this model, the suggested mechanism of action for these synthetic peptides is that they competitively bind to one of the heptad repeat regions when gp41 is in its extended conformation and prevent the structure from folding back onto itself. A 36-amino acid peptide (Fig. 23-2), corresponding to DP178 and later renamed T-20, pentafuside and finally marketed as ENF (Fuzeon), was found to be a potent inhibitor of HIV-1 in vitro, demonstrating a 50% inhibitory concentration (IC50) of 1.7 ng/mL in T cell lines.6
Preclinical Data
Because it is a peptide, ENF would be expected to undergo catabolism into individual amino acids already present in the body. However, detailed studies to determine the exact elimination of the drug are not available. In vitro experiments involving human hepatic tissues suggest that ENF initially undergoes hydrolysis to form a deamidated metabolite (abbreviated M3), and indeed this metabolite is detectable in human plasma soon after administration. In vitro studies suggest that ENF is not an inhibitor of CYP450 enzymes and does not appear to alter the metabolism of common substrates of CYP3A4, CYP2D6, and related hepatic enzymes. Conversely, drugs such as ritonavir and rifampin do not appear in preliminary studies to alter the pharmokinetic parameters of ENF in a clinically significant manner.14
ENF was not mutagenic or disruptive to chromosomes in a series of in vitro and animal studies. Long-term carcinogenicity studies have not been conducted. ENF had no effects on male or female fertility in rats at doses higher than the recommended human dose, relative to body surface area.14
PHASE I TRIAL
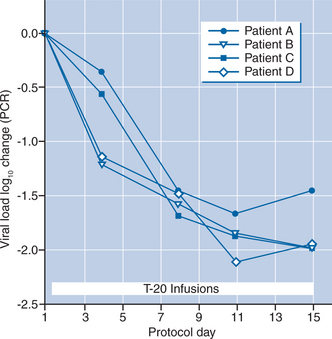
In the first clinical trial of a membrane fusion inhibitor,15 the ENF peptide was administered intravenously for 14 days to 16 HIV-infected adults. Patients received ENF monotherapy in a dose-escalation protocol, in which four patients each received 3, 10, 30, and finally 100 mg, all IV twice daily. No serious adverse effects were noted during short-term administration. The median half-life of the drug was 1.83 h. The nadir drug concentrations in the highest dose group were substantially higher than the IC50 of the drug and the overall pharmacokinetic profile suggested that intermittent or continuous subcutaneous T-20 administration might be feasible.
The viral load results demonstrated significant, dose-related declines in plasma HIV RNA levels during intravenous T-20 treatment. There was a significant decline in plasma HIV RNA when all 16 subjects were considered together (−0.39 log10; P < 0.05). The median viral load change in plasma viral load in the 100 mg dose group was −1.96 log10 by day 15 (Fig. 23-3). An analysis of viral dynamics showed that the initial slope of virus decline, a measure of antiretroviral potency, was comparable to that achieved with other approved HIV therapies including three and four drug combinations of RT and protease inhibitors (PIs). Thus, these findings provided “proof-of-concept” that therapeutics targeting a viral entry event could result in safe and clinically meaningful inhibition of viral replication.
PHASE II TRIALS
The second clinical trial of ENF (TRI-003) involved 78 subjects enrolled at multiple sites around the United States.16 This 28 day trial allowed patients to add ENF therapy to preexisting oral antiretroviral regimens. This was a dose-ranging study designed to compare continuous subcutaneous infusion (CSI) with intermittent subcutaneous injections of ENF, using escalating dose groups for each approach, in order to explore routes of administration more practical for the out-patient setting. Patients were eligible for the study if they had viral loads of >5000 copies/mL and were either on no other therapy or had not changed their antiretroviral regimen over the previous 6 weeks. Most subjects had advanced disease and extensive prior treatment histories with clinical resistance to many or all available RT and PIs, and thus the study was essentially measuring virologic responses to T-20 “functional monotherapy” in many cases.
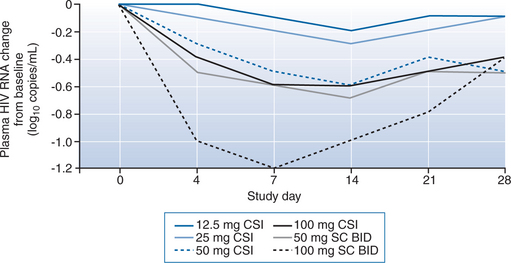
The CSI administration route proved problematic. The continuous infusion “insulin pump” devices frequently alarmed, suggesting that efficient flow was not achieved, and inferior trough drug concentrations in all CSI groups confirmed this. More subjects also experienced local injection site reactions (see adverse effects, below) in this group than the intermittent injection group. The concerns about the CSI administration route were partially alleviated by the pharmacokinetic results in the study, which suggested that continuous subcutaneous or intravenous administration were unnecessary since twice daily subcutaneous injections provided relatively stable drug levels for over 12 h after administration. Over a 28 day administration period, dose-related reductions in plasma HIV RNA levels were demonstrated that confirmed the findings in the phase I trial. The 100 mg CSI group experienced nearly a full order of magnitude (90%) decline, while the 50 mg twice daily and the 100 mg twice daily intermittent therapy groups had greater than 90% declines. The overall trend in the higher dose groups, however, was an initial decline followed by a more gradual return toward baseline viral loads (Fig. 23-4).
Sixty-one of the initial phase II subjects plus ten other subjects who had received prior ENF therapy were later enrolled into a 48-week rollover protocol. This protocol (T20-205) evaluated the long-term effects of administering open-label ENF, at 50 mg every 12 h subcutaneously, in addition to conventional, oral, highly active antiretroviral therapy (HAART) regimens.17 Investigators were provided with a genotypic analysis of each subject’s HIV isolate at baseline to help determine an “optimized background” (OB) HAART regimen for each individual. Overall, a mean viral load decline of 1.33 log10 copies/mL was achieved within 14 days of ENF plus HAART, and this degree of response was maintained through the remainder of the 48-week period (P < 0.001 for change from baseline at 48 weeks). The mean gain in absolute CD4 T-lymphocyte count at 48 weeks was 84.9 cells/μL (P < 0.001 for change from baseline). Potent responses were seen with comparable frequency regardless of the presence of multidrug resistant isolates, suggesting the possibility of a substantial fusion inhibitor contribution to the durability of response to the multiagent “salvage regimens”. Another phase II randomized pilot trial involved 71 patients who were protease-experienced but NNRTI-naive at entry.18 These subjects were randomized to a salvage antiretroviral regimen (abacavir, amprenavir, low dose ritonavir, and efavirenz) alone versus the same regimen plus one of three doses of subcutaneous ENF (50, 75, or 100 mg twice daily). Although the results were considered preliminary, there was a consistent dose-related trend toward additional virologic and immunologic benefit on ENF plus HAART. The pooled ENF arms achieved a median change of −2.27 log10 compared with −1.65 log10 in the control group. Interestingly, the differential response was greater among subjects with higher viral loads (>20 000 copies/mL) at baseline (median viral load change −2.64 log10 vs −1.55 log10). The gain in CD4+ T-lymphocyte counts over 16 weeks was +10 in the control group versus +64 and +74 in the 75 and 100 mg ENF arms, respectively.
PHASE III TRIALS
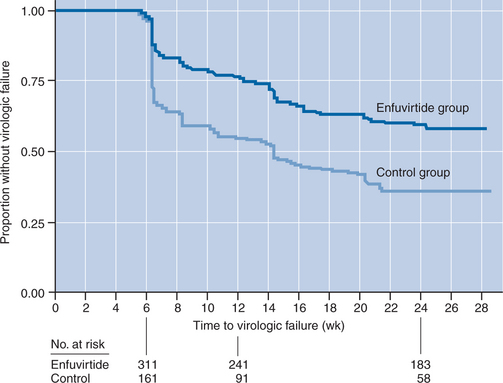
ENF received accelerated FDA approval based on the results of two large phase III randomized “T-20 Versus Optimized Regimen Only” trials (TORO 119 and TORO 220). Both trials evaluated the fusion inhibitor and OB; individualized regimens consisting of 3–5 drugs based on treatment histories, genotypic and phenotypic resistance testing) versus OB alone among patients with prior “triple class” (nucleoside RT inhibitors (NRTIs), nonnucleoside RT inhibitors (NNRTIs), and PIs) treatment experience. The two protocols combined enrolled ∼1000 subjects, TORO 1 (or T20-301) at sites in the Americas (US, Canada, Brazil) and TORO 2 (or T20-302) at sites in Europe and Australia. The two trials had nearly identical designs, randomizing treatment-experienced subjects 2:1 to ENF + OB versus OB alone, and the studies had very comparable results. Therefore, their results can reasonably be presented in parallel. The primary objective of these large trials was to determine whether there is an additional decline in viral load at 24 weeks and 48 weeks on ENF therapy beyond that seen with the optimized conventional therapeutic regimens alone.
Subjects were predominantly male (∼90% in both trials) and White (83% in TORO 1, 95% in TORO 2), with a median age of 42 years. Median baseline plasma viral loads and absolute CD4+ T-lymphocyte counts were >100 000 (5.1–5.2 log10) copies/mL and 75–100 cells/mm3, respectively. Most subjects (75–90%) had a prior history of an AIDS-defining event. Only a minority of subjects (∼20–25%) had viral isolates susceptible (by genotypic or phenotypic analyses) to three or more drugs in their chosen OB regimens, whereas a comparable proportion demonstrated in vitro evidence of resistance to all the OB agents they received. As expected in this context of extensive prior treatment, suboptimal clinical responses were quite common compared with typical protocols of newer agents involving treatment-naive subjects. There were significantly fewer protocol-defined cases of virologic failure between 8 and 24 weeks on the ENF-containing arms (41–48%) than the OB alone arms (63–76%) in both studies; failing subjects were provided access to open label ENF therapy. Thus, the intention-to-treat (ITT) analyses at 24 weeks and beyond likely underestimate advantages of the ENF-containing arms because of this crossover design. Nevertheless, the mean change from baseline viral load at 24 weeks was greater by nearly an order of magnitude in the ENF plus OB arm versus the OB alone arm in both protocols (−1.696 vs −0.764 in TORO 1 and −1.429 vs −0.648 in TORO 2, respectively; both differences P < 0.001). Whereas only a minority sustained viral load suppression below 400 copies/mL at 24 weeks overall. This favorable outcome was approximately twice as common in the arms receiving ENF plus OB versus OB alone (31.7% vs 16.4%, respectively, in TORO 1odds ratio = 3.17, P < 0.001); 28.4% vs 13.6%, respectively, in TORO 2 (odds ratio = 2.74; P < 0.001)). The time to protocol-defined failure also significantly favored the ENF arms in both studies (Fig. 23-5). Changes in absolute CD4+ T-lymphocyte counts were significantly greater for subjects receiving ENF plus OB versus OB alone in both studies (+76 vs +32 cells/mm3, respectively, in TORO 1; +65 vs +38 cells/mm3, respectively, in TORO 2; P < 0.02 for both differences). In longer term follow-up of TORO-1 and TORO-2 combined, at 48 weeks the proportion of subjects maintaining a viral load of <400 copies was 30% for ENF with OB versus 12% for OB alone.21 At 96 weeks, 26% of those remaining on ENF with OB continued to maintain a viral load <400; subjects who had delayed addition of ENF, after experiencing virologic failure on the protocol, were at a significant long-term disadvantage compared with those initially randomized to receive ENF with OB.22 Importantly, the achievement of >1 log decline in plasma viral load at 12 weeks on the TORO trials was a very strong predictor of beneficial responses at 24, 48, and 96 weeks of follow-up; subjects who did not experience this early response were quite unlikely to have sustained benefits (∼1–3%) at later time points.23
PEDIATRIC EXPERIENCE
There are limited published data regarding the efficacy of ENF in HIV-infected pediatric patients. Children’s Hospital Los Angeles investigators have reported on the long-term experience of 14 children (aged 4–12 years) who added on ENF 30–60 mg/m2 bid in the setting of incomplete HIV suppression on standard antiretroviral therapy (ART) regimens. Eleven of 14 had an initial substantial virologic response, and 10 subjects (70%) maintained >1 log improvement in viral load (allowing for changes in background regimens) at 24 weeks.24 Four subjects later experienced virologic failure (between weeks 40 and 63) in the open-label study. Of the original 14, six subjects still maintained virologic suppression >1 log below prestudy baseline at 96 weeks.25 Preliminary pharmacokinetic results from a study involving 25 children (aged 5–16) suggests that ENF can be dosed on a per weight basis (2 mg/kg) regardless of age (>5 years), body surface area or weight.26 The adverse experiences in these small protocols, predominantly local injection reactions, were generally comparable to the adult experience (summarized below).
PREGNANCY
It is not known whether ENF is excreted in human milk. When radiolabeled ENF was administered to lactating rats, radioactivity was detectable in the milk; it is unclear whether this radioactivity represents intact peptide or amino acid by-products of ENF metabolism. Regardless, due to the risks of HIV transmission and the unknown risks of ENF toxicity to the infant, mothers should be instructed not to breast-feed while receiving ENF.14
TOXICITY
Throughout phase I, II, and III ENF clinical trial experience, potential systemic reactions or internal organ damage associated with ENF were uncommonly reported; instead, most toxicity concerns have been related to local injection site reactions (ISRs; see Fig. 23-6). In the combined TORO 1/TORO 2 experience, the occurrence of some degree of ISR was nearly universal (∼98%), typically beginning during the first week of administration and not changing in severity over the period of continued administration.19 The majority of TORO subjects reported local induration (∼90%), erythema (∼90%), and nodular or cystic reactions (,75%). The majority of these local reactions were reported as mild (∼40–50%) or moderate pain (∼40–50%) at the injection site without limitation of usual activities. Less than 10% had limitations of usual activities or required nontopical analgesics for local discomfort, and none were hospitalized for ISRs. Less than 3% overall discontinued ENF due to ISRs during the first 24 weeks, and this figure remained <5% over more prolonged follow-up.27
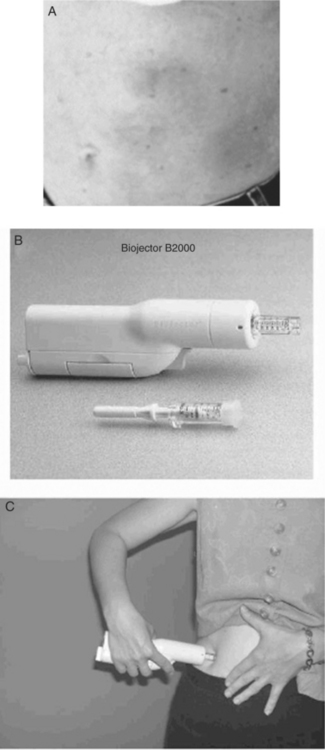
Biopsies of typical reaction sites reveal inflammation consistent with a local hypersensitivity reaction, in a pattern resembling granuloma annulare or an interstitial granulomatous drug reaction.28 Cutaneous biopsies of ISR lesions in the setting of more prolonged ENF administration (∼80 weeks) demonstrate increases in local T lymphocytes and some evidence of angiogenesis.29 Dermatologic findings may have variable clinical courses with different macroscopic patterns: transient infiltrative lesions, nodular lesions which resolve within 7–15 days, and sclerotic lesions persisting more than 30 days. Histologically, some lesions have an acute urticarial or vasculitic pattern with surrounding fat inflammation, whereas longer-lasting nodules may resemble scleroderma (but do not involve adnexal structures) and may slowly regress over time with rotation of injection sites. Anecdotal evidence suggests that some individuals benefit from adjuvant techniques aiming to avoid or diminish ISRs such as vigorously massaging the area immediately after injection, using smaller needles, or applying heat or ice to the site. However, data are limited to make any formal assessments regarding the risks and benefits of such manipulations.
Excluding ISRs, the overall rate of adverse events at 24 weeks in the TORO trials was roughly equivalent in the ENF with OB versus OB alone arms.19,20 The occurrence of severe adverse events was slightly higher in the ENF with OB arms, but there were no detectable trends in terms of specific categories or organ systems (essentially individual cases of various events typically attributable to ART regimens). Peripheral neuropathy and anorexia appeared slightly more common among ENF recipients, whereas diarrhea, nausea and vomiting were more commonly reported in those receiving OB alone.
Systemic “hypersensitivity reactions” to ENF are apparently uncommon (two cases or <1% of TORO subjects), but have been well-substantiated and may recur with rechallenge. One TORO subject had hypersensitivity manifesting as rash, fever, nausea and vomiting whereas the other had glomerulonephritis. One case report describes successful desensitization to ENF,30 although caution is probably warranted since one of the TORO subjects experienced severe respiratory distress during rechallenge. Whereas hypersensitivity reactions on the TORO protocols did not consistently correlate with increased circulating eosinophils, overall more subjects acquired eosinophilia in the ENF arms than in the OB only arms (∼11% vs ∼5%, respectively, developed >700 cells/mm3). There was no evidence that eosinophilia was associated with clinical signs or symptoms of hypersensitivity. In terms of other safety laboratory assessments in TORO 1 and 2, there were no meaningful trends distinguishing the Grade III or IV abnormalities observed in the ENV with OB versus the OB alone arms.
One unexpected finding from the combined TORO experience that has generated discussion was the higher incidence of bacterial pneumonia among ENF recipients (50 cases (5.6% or 4.9/100 patient years) on ENF with OB vs only one in the OB alone arms (90.3% or 0.6/100 patient-years); P = 0.02). There was also a higher incidence of overall bacterial infections and sepsis in the ENF with OB arms compared with OB alone arms. However, after adjustments for duration of exposure (since many subjects crossed over to open-label ENF, there were more monitored patient-years on ENF with OB than in the comparator arms), these more general infectious disease differences were not statistically significant. These trends have raised speculation about whether there are unanticipated effects of the membrane fusion peptide on immunologic responses or inflammatory pathways, or whether these pulmonary infections may relate somehow to chronic subcutaneous injections. On the other hand, the prevalence of pneumonia in either the ENF with OB or the OB only arms was lower than might be anticipated based on the historical experience of patients with similar stages of HIV infection, suggesting that ENF did not result in a dramatic increase over the background rate of respiratory infections. 48-week follow-up data on the TORO populations has confirmed the same trends of adverse events discussed above but with significantly fewer treatment-related adverse events overall in those originally randomized to receive ENF.27 Diarrhea, nausea and vomiting were slightly more common among those originally randomized to OB alone, whereas pneumonia and lymphadenopathy were conditions more common among ENF recipients. Quality of life assessments from the TORO trials demonstrated comparable outcomes regardless of treatment assignment,31 perhaps suggesting that the challenges of self-injections and ISRs were effectively offset by other subjective benefits. “General health” quality of life subcomponent scores improved more among ENF recipients than comparators, and mental health self-assessment scores were higher among ENF recipients than comparators at 24 weeks.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


