Endocrine Tumors
Samuel A. Wells Jr.
Over the past two decades, the remarkable advances in endocrine oncology have been enhanced by powerful and sophisticated molecular technology. In this regard studies of hereditary endocrine tumors have been especially fruitful, not only in defining the molecular genetic basis of these complex diseases, but in understanding the pathogenesis of the individual component tumor of the syndromes as well as their sporadic counterparts.
THE MULTIPLE ENDOCRINE NEOPLASIA SYNDROMES
Most endocrine tumors involve a single endocrine gland, are usually benign, and occur sporadically. Rarely, endocrine tumors occur in a familial pattern where they are multiple, involving more than one endocrine gland. Over the past 50 years at least six multiple endocrine neoplasia (MEN) syndromes have been described, including MEN-1, MEN-2, von Hippel-Lindau (VHL) disease, neurofibromatosis type 1, Carney complex (CNC), and the McCune-Albright syndrome (MAS). The genetic mutation causing each of these six MEN syndromes has been identified, in many cases leading to improved diagnosis and treatment of patients with a specific syndrome. It is not possible to address each of these syndromes in detail in this chapter, accordingly, the most common ones will be addressed.
Multiple Endocrine Neoplasia Type 1
Clinical Features
In 1954 Wermer1 described a family with hyperparathyroidism and tumors of the pancreatic islet cells and the pituitary gland. This hereditary syndrome has since been named multiple endocrine neoplasia (MEN) type 1 (MEN-1) (Mendelian Inheritance in Man [MIM] 13001) and over 20 separate endocrine or nonendocrine tumors may occur in patients with this disease. The prevalence of MEN-1 is 2 to 3 per 100,000, and males and females are equally affected. MEN-1 is characterized by high penetrance but variable expressivity. Virtually all patients develop parathyroid hyperplasia by age 40, 50% develop malignant pancreatic islet cell tumors (usually gastrinomas, less often insulinomas, and rarely glucagonomas, or vasoactive intestinal polypeptide secreting tumors [VIPomas]), and 25% develop pituitary tumors (usually prolactinomas). The diagnosis of MEN-1 is established either when a previously unaffected member of a family with MEN-1 develops a single characteristic endocrine tumor, or when a patient with hyperparathyroidism develops a pituitary tumor or a pancreatic islet cell tumor.
Molecular Genetics
Chandrasekharappa et al.2 discovered the genetic mutation for MEN-1. The MEN1 gene spans a 9.8 kb segment of chromosome 11q13 and consists of 10 exons with a 1,830 base pair region that encodes transcripts of 2.7 and 3.1 kb. The transcripts, expressed in almost all tissues, encode a novel, highly conserved 610 amino acid, 67 kDa protein, menin.3 MEN1, a putative tumor suppressor gene, mainly resides in the nucleus but is also found in the cytoplasm. The amino acid sequence of menin does not have homologies in the genome, thus clues to its mechanism of action have primarily come from identification of menin partnering to proteins or chromatin. Approximately 25 protein partners for menin have been identified, the first of which was junD, followed by others, such as MLL-containing complex, SMAD3, PEM, NM23, and nuclear factor B; however, at present no one menin partner has been shown convincingly to be critical to MEN-1 tumogenesis. The crystal structure of menin is unknown, and there is no direct evidence that menin binds directly to DNA; however, the protein appears to play a critical role in the regulation of gene transcription, apoptosis, and genome stability. Homozygous mice null for Men1 die during embryogenesis, while heterologous mice, Men1+/, develop a pattern of endocrine tumors very similar to that of patients with MEN-1.
The first “hit” in the MEN1 gene that leads to tumorigenesis involves small mutations in one or several bases, broadly distributed across the MEN1
open reading frame, such that half of newly diagnosed index cases are found to have novel mutations.4,5 The second “hit” is a chromosomal mutation in somatic tissue that causes frameshift (deletions, insertions, or splice site defects) and nonsense mutations, including a portion or all of the MEN1 gene. Approximately 75% of kindreds with MEN-1 will be found to have MEN1 mutations, however, in patients who demonstrate hyperparathyroidism and pituitary tumors, the incidence of MEN1 mutations is approximately 10%, suggesting another genetic cause of this MEN-1 variant. To date approximately 400 unique germline or somatic mutations of MEN1 have been described (Fig. 25.1).6 There is little correlation between genotype and phenotype, and presymptomatic diagnosis by direct DNA testing is useful only in identifying family members and then monitoring them for the development of specific endocrine tumors associated with MEN-1. There is no rationale for prophylactic removal of the parathyroid glands, the pancreas, or the pituitary gland in asymptomatic patients who have inherited a mutated MEN1 allele.
open reading frame, such that half of newly diagnosed index cases are found to have novel mutations.4,5 The second “hit” is a chromosomal mutation in somatic tissue that causes frameshift (deletions, insertions, or splice site defects) and nonsense mutations, including a portion or all of the MEN1 gene. Approximately 75% of kindreds with MEN-1 will be found to have MEN1 mutations, however, in patients who demonstrate hyperparathyroidism and pituitary tumors, the incidence of MEN1 mutations is approximately 10%, suggesting another genetic cause of this MEN-1 variant. To date approximately 400 unique germline or somatic mutations of MEN1 have been described (Fig. 25.1).6 There is little correlation between genotype and phenotype, and presymptomatic diagnosis by direct DNA testing is useful only in identifying family members and then monitoring them for the development of specific endocrine tumors associated with MEN-1. There is no rationale for prophylactic removal of the parathyroid glands, the pancreas, or the pituitary gland in asymptomatic patients who have inherited a mutated MEN1 allele.
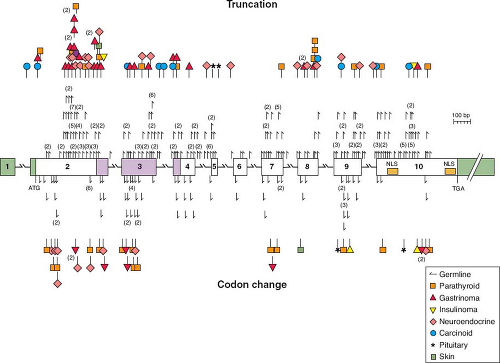 FIGURE 25.1 A schematic diagram of the genomic organization of the gene responsible for multiple endocrine neoplasia type 1 (MEN-1), including MEN-1 germline and somatic mutations. The gene contains ten exons (the first of which remains untranslated) and extends across 9 kb. It encodes a 610 amino acid protein, menin. Mutations shown above the exons cause menin truncation; those shown below the exons cause an amino acid or codon change. All unique mutations are represented; numbers in parentheses represent multiple reports of the same mutation in apparently unrelated individuals. The green-shaded areas indicate the untranslated regions. The location of the two nuclear localization signals (NLS), at codons 479-497 and 588-608, are indicated. Missense mutations in a region of menin (amino acids 139-242, identified by blue shading) prevented interaction with the AP1 transcription factor JunD. (From ref. 6, with permission.) |
The endocrine tumors characteristic of MEN-1 occur more commonly in a sporadic setting, where 25% of gastrinomas, 10% to 20% of insulinomas, 50% of VIPomas, 25% to 35% of bronchial carcinoids, and 20% of parathyroid adenomas express somatic MEN1 mutations. Conversely, somatic MEN1 mutations rarely if ever occur in sporadic adrenocortical tumors, pituitary tumors, or thyroid tumors.6
Other Hereditary Endocrinopathies Involving the Parathyroid and Pituitary Glands
The Parathyroid Gland
While familial hyperparathyroidism is most commonly a component of MEN-1, it can occur in
other settings, the most notable being familial (benign) hypocalciuric hypercalcemia (FHH), the hyperparathyroidism jaw-tumor (HPT-JT) syndrome, familial isolated hyperparathyroidism (FIHP), and MEN-2A.
other settings, the most notable being familial (benign) hypocalciuric hypercalcemia (FHH), the hyperparathyroidism jaw-tumor (HPT-JT) syndrome, familial isolated hyperparathyroidism (FIHP), and MEN-2A.
Familial Hypocalciuric Hypercalcemia (FHH, MIM 145980).
The calcium-sensing receptor (CASR), a critical regulator of extracellular calcium homeostasis, is a seven-transmembrane-spanning G-protein-coupled receptor, which is expressed in cells of the parathyroid gland and the kidney tubule. The discovery of the CASR was a surprise, since previously no small cation had been shown capable of acting as a ligand for a G-protein-coupled receptor.7 The human CASR, encoded by six exons of the gene located on chromosome 3q113.3-q21, is sensitive to changes in the ambient calcium concentration and when activated it inhibits parathyroid hormone (PTH) secretion and the renal reabsorption of calcium. With heterogenous inactivating mutations of the CASR gene the parathyroid cell fails to sense properly an increased serum calcium concentration, and the resulting increase in PTH secretion causes FHH, an autosomal dominant disease characterized by hypocalciuria, hypercalcemia, and parathyroid hyperplasia. It is important for clinicians to recognize this relatively mild form of familial hyperparathyroidism; although parathyroidectomy reduces the serum calcium level, it is only temporary. With inactivating mutations in both alleles of the CASR gene, neonatal severe hyperparathyroidism ([NSHPT] MIM 239200) develops with serum calcium levels in the range of 15 to 20 mg/dL. This disease represents a life-threatening emergency and urgent parathyroidectomy is indicated.8 Conversely, activating mutations of the CASR gene cause the parathyroid cells to sense that serum calcium is “elevated” when it is actually normal. There is a resulting decrease in the blood calcium level expressed as the syndrome of autosomal dominant hypoparathyroidism ([ADH] MIM 168468).9 To date approximately 115 mutations (60% inactivating and 40% activating) have been described in the CASR gene, and most are missense mutations clustered in exons 3, 4, and 7.
Hyperparathyroidism-Jaw Tumor Syndrome (HPT-JT, MIM 145001).
HPT-JT is characterized by the autosomal dominant occurrence of hyperparathyroidism, ossifying fibromas of the mandible or maxilla, renal cysts or solid tumors, and uterine fibromas.10 Approximately 50 families with HPT-JT have been reported and 80% of patients have hyperparathyroidism, and in 15% of cases the parathyroid tumors are malignant. It is noteworthy that the age-related penetrance of HPT-JT is approximately 40% by age 40 in contrast to MEN-1 where the age-related penetrance is 98% by age 40.11 Members of kindreds with this disease need lifelong surveillance by physical examination and biochemical evaluation. It has even been suggested that serial ultrasound examination of the neck should be performed as parathyroid carcinoma has been reported in normocalcemic kindred members.12
The cause of HPT-JT appears to be a tumor suppressor gene, HRPT2, which is located on chromosome 1q25-q31. The HPRT2 gene consists of 17 exons and the mutations are scattered throughout the 1593 coding region, with the majority resulting in functional loss through premature truncation.13 Approximately 16 activating mutations of HRPT2, most of which are frameshift, have been identified. The HPRT2 gene encodes a 531 amino acid protein, parafibromin (named for parathyroid tumors and jaw fibromas).14 The function of parafibromin is unknown but it is thought to regulate posttranscriptional events and histone modification. There is recent evidence that parafibromin has pro-apoptotic activity, important as a tumor suppressor function.15 Germline mutations in HPRT2 have been identified in approximately half of HPT-JT families, and even in some families with MEN-2A.
Besides the evidence of germline mutations in patients with the HPT-JT somatic HRPT2 mutations have been detected in the majority of patients with sporadic parathyroid carcinoma.16,17 It is important to note that mutations in HRPT2 are rarely if ever seen in sporadic parathyroid adenomas, an important diagnostic finding distinguishing benign from malignant parathyroid tumors.15,18
Familial Isolated Hyperparathyroidism (FIHP, MIM 145000).
The syndrome of FIHP is a heterogenous condition, and some kindreds thought to have this disease have been shown to have germline mutations of MEN1, CASR, or HRP-JT, suggesting that the disease represents incompletely expressed forms of MEN1, FHH, or HPT-JT.19 Over 100 families with FIHP have been reported and in most cases the causative genetic mutation is unknown, although there are convincing data that it resides on chromosome 2p13.3-14.20
The Pituitary Gland
There is no relationship between genotype and phenotype in patients with MEN-1, thus there is no way to predict the presence or behavior of pancreatic islet cell tumors or pituitary tumors. There are, however, important genetic mutations associated with other hereditary and sporadic pituitary tumors.
Carney Complex (CNC, MIM 160980).
Pituitary adenomas can occur as a component of CNC, a familial disease characterized by hypersomatotropenemia, cardiac or cutaneous myxomas, spotty skin pigmentation, primary pigmented nodular adrenal disease, and testicular tumors.21 About
75% of patients exhibit subclinical increases in growth hormone (GH), insulinlike growth factor-1 (IGF-1), and prolactin. Acromegaly occurs in about 10% of patients. Genetic linkage analysis has shown two loci for CNC, one on chromosome 2p16 (CNC2), and the other on chromosome 17q22-24 (CNC1). Neither locus is associated with a specific phenotype. In more than 50% of cases the CNC has been linked to an inactivating mutation in the gene coding for the protein kinase A (PKA) type 1α subunit, PRKAR1A, at 17q24. The 2p16 locus is uncharacterized.
75% of patients exhibit subclinical increases in growth hormone (GH), insulinlike growth factor-1 (IGF-1), and prolactin. Acromegaly occurs in about 10% of patients. Genetic linkage analysis has shown two loci for CNC, one on chromosome 2p16 (CNC2), and the other on chromosome 17q22-24 (CNC1). Neither locus is associated with a specific phenotype. In more than 50% of cases the CNC has been linked to an inactivating mutation in the gene coding for the protein kinase A (PKA) type 1α subunit, PRKAR1A, at 17q24. The 2p16 locus is uncharacterized.
Familial Isolated Pituitary Adenomas (FIPA, MIM 102200).
Pituitary adenomas can also occur as FIPA. Daly et al.22 evaluated 64 families with FIPA, residing in Belgium, France, Italy, and the Netherlands. Of the 138 affected family members, 55 had prolactinomas, 47 had somatotropinomas, 28 had nonsecreting adenomas, and 8 had adrenocorticotropic hormone-secreting tumors. The incidences of a homogenous (single tumor) phenotype and a heterogeneous tumors phenotype were approximately equal. Affected patients were found to have no mutations in either the MEN1 or PRKRA1A genes.
Recently, Vierimaa et al.,23 in a study of cases of low penetrance familial pituitary adenoma in Northern Finland identified loss of mutation functions in the aryl hydrocarbon receptor interacting protein (AIP) gene. AIP forms a complex with the aryl hydrocarbon receptor (AHR) and two 90-kD heat-shock proteins (HSP90). AHR is a ligand-activated transcription factor and also participates in cellular signaling pathways.
Expression profiling in two families resulted in the identification of AIP as one of the candidate genes, and a nonsense mutation (Gln14X) was also identified and found to segregate with the GH-secreting adenomas.23 Daly et al.24 studied 73 FIPA families with 156 individuals and found that 11 of them harbored 10 AIP germline mutations. Kindred members with AIP mutations, compared to those without mutations, were younger and had larger tumors. Growth hormone producing tumors predominated among family members with AIP mutations.
Multiple Endocrine Neoplasia Type 2
Clinical Features
In 1968 Steiner et al.25 described a large family with medullary thyroid carcinoma (MTC), pheochromocytomas, hyperparathyroidism, and Cushing’s syndrome. The disease was named multiple endocrine neoplasia type 2, and it is now recognized that there are three related syndromes characterized by hereditary MTC: MEN-2A (MIM 171400), MEN-2B (MIM 162300), and familial medullary thyroid carcinoma (FMTC) (MIM 155240). There is near complete penetrance but variable expressivity, as virtually all patients with MEN-2A develop MTC, approximately half develop pheochromocytomas, and 30% develop hyperparathyroidism. Less often MEN-2A is associated with cutaneous lichen amyloidosis (CLA), which develops on the upper back and serves as a precocious marker of the disorder, or Hirschsprung’s disease (HD), which is characterized by loss of ganglion cells in variable segments of the large bowel. Patients with MEN-2B develop MTC and pheochromocytomas with the same frequency as patients with MEN-2A, but rather than hyperparathyroidism, they express a generalized gastrointestinal neuromatosis and a characteristic physical appearance. Patients with FMTC develop only MTC. Of patients with hereditary MTC, 80% have MEN-2A, 15% have FMTC, and 5% have MEN-2B.
Medullary thyroid carcinoma originates from C cells, which are derived from the neural crest. The C cells have great biosynthetic capability and secrete the polypeptide hormone calcitonin (CTN) and the glycoprotein carcinoembryonic antigen (CEA). Plasma CTN serves as an excellent marker for MTC, and presently its main use is in detecting persistent or residual MTC following thyroidectomy.
The MTC is the most common cause of death in patients with MEN-2A, MEN-2B, and FMTC, and the only effective therapy is timely thyroidectomy as standard chemotherapy and external beam radiotherapy are not useful.
Molecular Genetics
In 1985 Takahashi et al.26 discovered the RET (REarranged during Transfection) protooncogene. The gene is located in the pericentromeric region of chromosome 10q11.2 and includes 21 exons. RET encodes a receptor tyrosine kinase, which is expressed in neuroendocrine cells (including thyroid C cells and adrenal medullary cells), neural cells (including parasympathetic and sympathetic ganglion cells), urogenital tract cells, and the branchial arch cells.
The RET gene has an extracellular portion, which contains four cadherin-like repeats, a calcium binding site and a cysteine-rich region, a transmembrane portion, and an intracellular portion, which contains two tyrosine kinase domains. Alternate splicing of RET produces three isoforms with either 9, 43, or 51 amino acids at the C terminus, referred to as RET9, RET43, and RET51.27,28 Mice lacking RET51 are normal, however, mice lacking RET9 have renal malformation and defects in innervation of the gut.29
RET is essential for the development, survival, and regeneration of many neuronal cells in the gut, the kidney, and the nervous system. A tripartite complex is necessary for RET signaling. One of
four glial-derived neurotrophic factors (GDNF) family ligands (GFLs)—GDNF, neurturin, persephin, or artemin-binds RET in conjunction with one of four glycosylphosphatidylinositol-anchored coreceptors, designated GDNF family receptors (GFR): GFR-α1, GFR-α2, GFR-α3, or GFR-α4.30,31,32 The GFL-GFR complex causes dimerization of two RET molecules with activation of autophosphorylation and intracellular signaling. The C-terminal of RET contains 16 tyrosine residues, among which Y905 is a binding site for Grb7/10 adaptors, Y1015 a binding site for phospholipase Cγ, Y981 a binding site for c-Src, and Y1096 a binding site for Grb2. Tyrosine 1072 is a multidocking binding site for such proteins as SHC, SHCC, IRS1/2, FRS2, DOK1/4/5/, and Enigma. The RET receptor may activate various signaling pathways through Y1072, which thereby serves as a prerequisite for initiating transformation of RET-derived oncogenes in cell cultures and transgenic animals.33




four glial-derived neurotrophic factors (GDNF) family ligands (GFLs)—GDNF, neurturin, persephin, or artemin-binds RET in conjunction with one of four glycosylphosphatidylinositol-anchored coreceptors, designated GDNF family receptors (GFR): GFR-α1, GFR-α2, GFR-α3, or GFR-α4.30,31,32 The GFL-GFR complex causes dimerization of two RET molecules with activation of autophosphorylation and intracellular signaling. The C-terminal of RET contains 16 tyrosine residues, among which Y905 is a binding site for Grb7/10 adaptors, Y1015 a binding site for phospholipase Cγ, Y981 a binding site for c-Src, and Y1096 a binding site for Grb2. Tyrosine 1072 is a multidocking binding site for such proteins as SHC, SHCC, IRS1/2, FRS2, DOK1/4/5/, and Enigma. The RET receptor may activate various signaling pathways through Y1072, which thereby serves as a prerequisite for initiating transformation of RET-derived oncogenes in cell cultures and transgenic animals.33
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
