div class=”ChapterContextInformation”>
8. The Endocrine Pancreas and Diabetes Mellitus
8.1 The Pancreas
the exocrine pancreas, the major digestive gland of the body, and
the endocrine pancreas, the source of insulin, glucagon, somatostatin, pancreatic polypeptide (PP), and ghrelin.
Whereas the major role of the products of the exocrine pancreas is the processing of ingested foodstuffs (← the digestive enzymes) so that they become available for absorption, the hormones of the endocrine pancreas modulate every other aspect of cellular nutrition from rate of adsorption of foodstuffs to cellular storage or metabolism of nutrients.
8.1.1 Anatomy and Histology
The endocrine pancreas consists of approximately one million small endocrine glands—the islets of Langerhans—scattered throughout the glandular substance of the exocrine pancreas, comprising 1–1.5% of the total mass of the pancreas. At least five cell types—α, β, D (δ), ε, and PP—have been identified in the islets. Each of these islet cell types produces a distinguishing peptide hormone: glucagon, insulin, somatostatin, ghrelin, and PP, respectively.
8.1.2 Hormones of the Endocrine Pancreas
8.1.2.1 Insulin
Biosynthesis
The human insulin gene resides on the short arm of chromosome 11. A unique set of transcription factors found in the β cell nucleus activates the transcription of the preproinsulin mRNA from the insulin gene, resulting preproinsulin, which then is cleaved to proinsulin by microsomal enzymes. Further, proinsulin is converted into insulin and a smaller connecting peptide, or C peptide, by proteolytic cleavage at two sites along the peptide chain, a process that takes place into the Golgi apparatus.
Biochemistry
Proinsulin consists of a single chain of 86 amino acids, which includes the A and B chains of the insulin molecule plus a connecting segment of 35 amino acids. The prohormone-converting enzymes type 1 and 2 recognize and cut at pairs of basic amino acids, thereby removing the intervening sequence. After the two pairs of basic amino acids are removed by carboxypeptidase E, the result is a 51 amino acid insulin molecule and a 31 amino acid residue, the C peptide. A small amount of proinsulin escapes cleavage and is secreted intact into the bloodstream, along with insulin and C peptide.
C peptide, the 31 amino acid peptide, released during cleavage of insulin from proinsulin in equimolar amounts with insulin, has no known biologic activity.
Insulin is a protein consisting of 51 amino acids contained within two peptide chains: an A chain, with 21 amino acids; and a B chain, with 30 amino acids. The chains are connected by two disulfide bridges. In addition, an intrachain disulfide bridge links positions 6 and 11 in the A chain. Endogenous insulin has a circulatory half-life of 3–5 min.
Secretion
The human pancreas secretes about 30 units of insulin per day into the portal circulation of normal adults in distinct pulses with a period of approximately 5 min. The basal concentration of insulin in the peripheral blood of fasting humans averages 10 μU/mL (0.4 ng/mL or 61 pmol/L). In normal control subjects, insulin seldom rises above 100 μU/mL (610 pmol/L) after standard meals. After ingestion of food, peripheral insulin concentration increases within 8–10 min, reaches peak concentrations by 30–45 min, and then rapidly declines to baseline values by 90–120 min postprandially.
Basal insulin secretion occurs in the absence of exogenous stimuli, in the fasting state. Plasma glucose levels below 80–100 mg/dL (4.4–5.6 mmol/L) do not stimulate insulin release. Stimulated insulin secretion occurs in response to exogenous stimuli. Glucose is the most potent stimulant of insulin release. The perfused pancreas releases insulin in two phases: when the glucose concentration increases suddenly, an initial short-lived burst of insulin release occurs (the first phase); if the glucose elevation persists, the insulin release gradually falls off and then begins to rise again to a steady level (the second phase). However, sustained levels of high glucose stimulation (≈4 h in vitro or >24 h in vivo) result in a reversible desensitization of the β-cell response to glucose but not to other stimuli.
The β-cell senses glucose through its metabolism. Glucose enters the pancreatic β cell by passive diffusion, facilitated by membrane proteins termed glucose transporters (GLUTs) (see later). The low-affinity enzyme glucokinase catalyzes the subsequent, and rate-limiting, step in glucose metabolism by the pancreatic β cell, the phosphorylation of glucose to glucose-6-phosphate. Glucose catabolism in the β cell causes a ↑ in the intracellular ATP-ADP ratio. Acting through the sulfonylurea receptor (SUR1), the nucleotide-sensing subunit of the ATP-sensitive potassium channels on the surface of the β cell, the rise in ATP:ADP ratio closes the potassium channels and depolarizes the cell, thereby activating the voltage-sensitive calcium channels and allowing the entry of calcium ions into the cell.
Insulin release requires calcium ion signaling. Some nonglucose stimuli of insulin release also function through increases in cytoplasmic Ca2+. The sulfonylurea and meglitinide (such as repaglinide) medications act by closing the ATP-sensitive potassium channels.
Glucose metabolism in the β cell also generates additional signals that amplify the secretory response to elevations in cytoplasmic Ca2+ concentration. The exact mechanisms of these amplifying signals remain unknown but involve multiple pathways and include increases in the intracellular signaling molecules diacylglycerol and cyclic adenosine monophosphate (cAMP).
Insulin Receptors and Insulin Action
Insulin action begins with the binding of insulin to a receptor on the surface of the target cell membrane. Most cells of the body have specific cell surface insulin receptors.
Metabolic Effects of Insulin
The major function of insulin is to promote storage of ingested nutrients.
Paracrine Effects
Paracrine effects of the β and δ cells on the nearby α cells are of considerable importance in the endocrine pancreas. Insulin directly inhibits α cell secretion of glucagon. In addition, somatostatin, released by δ cells in response to most of the same stimuli that provoke insulin release, also inhibits glucagon secretion.
Because glucose stimulates only β and δ cells (whose products then inhibit α cells) whereas amino acids stimulate glucagon as well as insulin, the type and amounts of islet hormones released during a meal depend on the ratio of ingested carbohydrate to protein. The higher the carbohydrate content of a meal, the lower the amount of glucagon released by any amino acids absorbed. In contrast, a predominantly protein meal results in relatively greater glucagon secretion.
Endocrine Effects
Liver
Insulin promotes anabolism—Insulin promotes glycogen synthesis and storage while inhibiting glycogen breakdown. These effects are mediated by changes in the activity of enzymes in the glycogen synthesis pathway (see below). Insulin increases both protein and triglyceride synthesis and very low-density lipoprotein (VLDL) formation by the liver. It also inhibits gluconeogenesis and promotes glycolysis through its effects on the function and expression of key enzymes of both pathways.
Insulin inhibits catabolism—Insulin acts to reverse the catabolic events of the postabsorptive state by inhibiting hepatic glycogenolysis, ketogenesis, and gluconeogenesis.
Muscle
Insulin promotes protein synthesis in muscle by increasing amino acid transport, as well as by stimulating ribosomal protein synthesis. In addition, insulin promotes glycogen synthesis to replace glycogen stores expended by muscle activity. This is accomplished by increasing glucose transport into the muscle cell, enhancing the activity of glycogen synthase, and inhibiting the activity of glycogen phosphorylase.
Adipose Tissue
Insulin acts to promote triglyceride storage in adipocytes by a number of mechanisms. (1) It induces the production of lipoprotein lipase in adipose tissue (this is the lipoprotein lipase that is bound to endothelial cells in adipose tissue and other vascular beds), which leads to hydrolysis of TGs from circulating lipoproteins, thereby yielding free fatty acids (FFAs) for uptake by adipocytes. (2) By ↑ glucose transport into fat cells, insulin increases the availability of α-glycerol phosphate, a substance used in the esterification of FFAs into TGs. (3) Insulin inhibits intracellular lipolysis of stored TGs by inhibiting intracellular lipase (also called hormone-sensitive lipase). This reduction of fatty acid flux to the liver is a key regulatory factor in the action of insulin to lower hepatic gluconeogenesis and ketogenesis.
Central Nervous System
Insulin signaling via PI3 kinase in key cells in the hypothalamus functions with leptin signaling to ↓ appetite and ↑ energy expenditure.
Glucose Transporter Proteins
Glucose oxidation provides energy for most cells and is critical for brain function. Because cell membranes are impermeable to hydrophilic molecules such as glucose, all cells require carrier proteins to transport glucose across the lipid bilayers into the cytosol. Whereas the intestine and kidney have an energy-dependent Na+-glucose cotransporter, all other cells utilize non-energy-dependent transporters that facilitate diffusion of glucose from a higher concentration to a lower concentration across cell membranes. Facilitative glucose transporters comprise a large family including at least 13 members, with the first four members of the family (GLUT-1, GLUT-3, GLUT-2, and GLUT-4) being the best characterized. They have distinct affinities for glucose and distinct patterns of expression.
8.1.2.2 Glucagon
Biochemistry
Pancreatic α cells generates → the glucagon peptide from the large proglucagon peptide encoded by the preproglucagon gene located on human chromosome 2.
Secretion
In contrast to its stimulation of insulin secretion, glucose inhibits glucagon secretion. Conflicting data surround the question of whether glucose directly inhibits secretion from the α cell or whether it only acts via release of insulin and somatostatin from the β and δ cells, both of which inhibit the α cell directly.
Action of Glucagon
In contrast to insulin, glucagon signaling in the liver stimulates the breakdown of stored glycogen, maintains hepatic output of glucose from amino acid precursors (gluconeogenesis), and promotes hepatic output of ketone bodies from fatty acid precursors (ketogenesis). Glucagon facilitates the uptake of the gluconeogenic substrate alanine by liver and directs fatty acids away from re-esterification to TGs and toward ketogenic pathways.
⇒ glucagon signaling results in the net release of readily available energy stores from the liver in the form of glucose and ketones to the other tissues between meals.
8.1.2.3 Somatostatin
Pancreatic δ cells → a 14 amino acid cyclic polypeptide, somatostatin-14. First identified in the hypothalamus, somatostatin has been found in a number of tissues, including many areas of the brain and peripheral nervous system, the endocrine D cells in the epithelial lining of the stomach and intestine, and the cells in the pancreatic islets.
! Somatostatin-28 is 10 times more potent than somatostatin-14 in inhibiting GH and insulin secretion, whereas somatostatin-14 is more effective in inhibiting glucagon release.
8.1.2.4 Pancreatic Polypeptide
PP ← PP cells located chiefly in islets in the posterior portion of the head of the pancreas.
Although it has been implicated in the regulation of exocrine pancreatic secretion and gall bladder contraction, the physiological actions of PP remain uncertain.
↑ values in: pancreatic endocrine tumors such as glucagonoma or vasoactive intestinal polypeptide-secreting tumor and in all tumors of the pancreatic PP cell.
8.1.2.5 Ghrelin
⊕ GH secretion directly through its receptor on pituitary somatotrophs, and also through its stimulation of hypothalamic GHRH secretion.
induces gastric emptying and acid secretion and regulates appetite and energy balance via neurons in the arcuate nucleus of the hypothalamus.
8.2 Diabetes Mellitus
8.2.1 Background
Diabetes mellitus (DM) is a metabolic disorder of multiple etiology characterized by chronic hyperglycemia with disturbances of carbohydrate, fat, and protein metabolism due to an absolute or relative deficiency of insulin secretion and/or insulin action (insulin resistance (IR)). Worldwide, 285 million people were diagnosed with diabetes in 2010, and this number is predicted to exceed 400 million by 2030.
DM is associated with development of specific long-term organ damage (diabetes complications) including retinopathy with potential blindness, nephropathy with a risk of progression to renal failure, neuropathy with risk for foot ulcers, amputation, autonomic dysfunction such as sexual impairment.
Patients with diabetes are at a particularly high risk for cardiovascular, cerebrovascular, and peripheral artery disease.
Associated with ↓ life expectancy, quality of life.
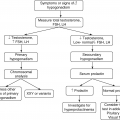
8.2.2 Diagnosis
Definition of glycemic “status”
• Normoglycemia |
Fasting plasma glucose (FPG) <110 mg/dL 2-h PPG (postprandial 2 h, OGTT) <140 mg/dL |
• Intermediate hyperglycemia: |
Impaired glucose tolerance (IGT) FPG < 126 mg/dL (7.0 mmol/L) and 2-h plasma glucose (OGTT): ≥140 < 200 mg/dL) (≥7.8 and <11.1 mmol/L) |
Impaired fasting glucose (IFG) FPG: 110–125 mg/dL (6.1–6.9 mmol/L) and 2-h plasma glucose (OGTT): <140 mg/dL (7.8 mmol/) |
• Diabetes mellitus |
FPG ≥ 126 mg/dL (7.0 mmol/L)a OR 2-h plasma glucose ≥200 mg/dL (11.1 mmol/L) during OGTT. The test should be performed as described by the WHO, using a glucose load containing the equivalent of 75-g anhydrous glucose dissolved in watera OR A1c ≥ 6.5% (48 mmol/mol) OR In a patient with classic symptoms of hyperglycemia or hyperglycemic crisis, a random plasma glucose ≥200 mg/dL (11.1 mmol/L) |
Criteria for testing for diabetes or prediabetes in asymptomatic adults (ADA 2018) |
|---|
1. Testing should be considered in overweight or obese (body mass index [BMI] ≥25 kg/m2 or ≥23 kg/m2 in Asian Americans) adults who have one or more of the following risk factors: |
First-degree relative with diabetes High-risk race/ethnicity (e.g., African American, Latino, Native American, Asian American, Pacific Islander) History of cardiovascular disease (CVD) Hypertension (≥140/90 mmHg or on therapy for hypertension) High-density lipoprotein (HDL)-cholesterol level <35 mg/dL (0.90 mmol/L) and/or a TG level >250 mg/dL (2.82 mmol/L) Women with polycystic ovary syndrome (PCOS) Physical inactivity Other clinical conditions associated with IR (e.g., severe obesity, acanthosis nigricans) |
2. Patients with prediabetes (A1c > 5.7% [39 mmol/mol], IGT, or IFG) should be tested yearly |
3. Women who were diagnosed with gestational diabetes mellitus (GDM) should have lifelong testing at least every 3 years |
4. For all other patients, testing should begin at the age of 45 years |
5. If results are normal, testing should be repeated at a minimum of 3-year intervals, with consideration of more frequent testing depending on initial results and risk status |
8.2.3 Classification of Diabetes Mellitus (Table 8.2)
Etiological classification of diabetes mellitus
1. Type 1 diabetes (due to autoimmune β-cell destruction, usually leading to absolute insulin deficiency) (5–10% of cases) (a) Includes autoimmune, type 1A and idiopathic forms, type 1B |
2. Type 2 diabetes (due to a progressive loss of β-cell insulin secretion frequently on the background of insulin resistance) (90–95% of cases) |
3. Gestational diabetes mellitus (GDM) (diabetes diagnosed in the second or third trimester of pregnancy that was not clearly overt diabetes prior to gestation) |
4. Specific types of diabetes due to other causes (a) Monogenic diabetes Autosomal dominant genetic defects of B-cell function: • Maturity onset diabetes of the young (MODY) Other genetic defects of B-cell function • Mitochondrial DNA mutations Neonatal diabetes Genetic defects of insulin action: • Lipodystrophies • Insulin receptor mutations (b) Other genetic syndromes sometimes associated with diabetes (e.g., Down syndrome, Friedreich’s ataxia, Klinefelter syndrome, Wolfram syndrome, Lawrence–Moon–Biedl syndrome, myotonic dystrophy, Prader–Willi syndrome, others) (c) Secondary diabetes Diseases of the exocrine pancreas: • Pancreatitis, cystic fibrosis, trauma, pancreatectomy, neoplasia, pancreatic destruction (including cystic fibrosis and hemochromatosis), others. Endocrinopathies: • Cushing syndrome, acromegaly, pheochromocytoma, glucagonoma, somatostatinoma, hyperthyroidism, aldosteronoma Drug- or chemical-induced: • Glucocorticoids, thyroid hormone, diazoxide, β-adrenergic agonists, thiazides, G-interferon, antiretroviral treatment (HIV) Infections: • Congenital rubella, cytomegalovirus (CMV), others |
Prediabetes-defied as IFG or IGT |
8.2.4 Diabetes Pathogenesis
8.2.4.1 Type 1 DM
Type 1 DM is immune-mediated in >95% of cases (type Ia) and idiopathic in <5% (type Ib).
Immune-Mediated Diabetes
commonly occurs in childhood and adolescence, but it can occur at any age, even in the eighth and ninth decades of life.
Autoimmune destruction of β cells resulting in impaired insulin secretion has multiple genetic predispositions and is also related to environmental factors that are still poorly defined.
Risk Factors
Personal history of other autoimmune diseases including Graves disease, myasthenia gravis, celiac disease, and pernicious anemia; family history of autoimmune diseases.
Genetics of Type 1 DM
The disease has strong the human leukocyte antigen (HLA) associations, with linkage to the DQA and DQB genes. The HLA-DR/DQ alleles can be either predisposing or protective. The HLA class II DR and DQ loci (respectively encoded by genes DRB and DQB) account for almost all type 1 DM susceptibility from this region and >90% of patients with type 1 DM carry at least one copy of the DRB*301-DQB*201 or DRB*401-DQA*301-DQB*302 (allele numbers after asterisk). Other susceptibility loci include variants in the gene encoding insulin (INS) that confers a twofold increased risk, etc. Both candidate gene studies and genomic-wide association studies (GWAS) have identified a number of additional risk loci that make smaller contribution to the genetic risk of type 1 DM.
Environmental Factors in Type 1 DM
While genetic inheritance may play an important role in causing type 1 DM, the monozygotic twin studies demonstrate that other causes, such as environmental, are at least as important. Most individuals with type 1 DM do not have other family members with the disease. Environmental factors associated with increased risk of type 1 DM include viruses (mumps, congenital rubella, Coxsackie virus B4), cow’s milk proteins, toxic chemical agents, or destructive cytotoxins such as hydrogen cyanide.
Direct damage to β cells
Initiators or accelerators of the autoimmune attack on the β cells
Molecular mimicry (the immune system mistakenly targets β-cell proteins that share homologies with certain viral or other foreign peptides)
Islet cell autoantibodies (ICA)
Autoantibodies to GAD (GAD65)
Insulin (IAA)
The tyrosine phosphatases IA-2 (ICA512) and IA-2b
Zinc transporter 8 (ZnT8) antibodies
Although useful for diagnosis and predicting type 1 DM, antibodies against β-cell proteins do not directly cause the destruction of β cells in type 1DM. Instead, it is the cellular immune system, the T lymphocytes that infiltrate the islet (the process is called insulitis) and destroy the β cells. At the time of diagnosis, the islets are extensively infiltrated with T lymphocytes. Normally, the thymus detects autoreactive T cells during development so that the immune system becomes tolerant of self-antigens. In addition, certain specialized T cells, the regulatory T cells, further prevent attacks against healthy tissues by restraining the activity of any autoreactive T cells that escape the thymus. Type 1 DM results from a breakdown in the processes of self-tolerance in the immune system. Eighty percent of β cell mass is destroyed before features of DM present. After initial presentation, honeymoon period often occurs where glycemic control can be achieved with little or no insulin treatment as residual cells are still able to produce insulin. Once these cells are destroyed, there is complete insulin deficiency.
The rate of β-cell destruction is quite variable, being rapid in some individuals (mainly infants and children) and slow in others (mainly adults). At the later stage of the disease, there is little or no insulin secretion, as manifested by low or undetectable levels of plasma C-peptide. Type 1 DM is a catabolic disorder in which circulating insulin is virtually absent, plasma glucagon is ↑ and the pancreatic β cells fail to respond to all known insulinogenic stimuli. In the absence of insulin, the main target tissues of insulin—liver, muscle, and fat—not only fail to appropriately take up absorbed nutrients but continue to deliver glucose, amino acids, and fatty acids into the circulation from their respective storage depots. Furthermore, alterations in fat metabolism lead to the production and accumulation of ketones. This inappropriate persistence of the fasted state postprandially can be reversed by administration of insulin.
Type 1b Diabetes
Approximately, 5% of patients with the clinical features of type 1 DM lack serum evidence of autoimmunity. This probably represents a heterogeneous group of disorders that lead to profound β-cell destruction or loss, absolute insulin deficiency, and a syndrome clinically similar to autoimmune type 1a DM.
8.2.4.2 Type 2 DM
It occurs more frequently in:
women with prior GDM,
those with hypertension or dyslipidemia, and
certain racial/ethnic subgroups (African American, American Indian, Hispanic/Latino, and Asian American).
Etiology
Complex and multifactorial.
↔ often associated with a strong genetic predisposition or family history in first-degree relatives, more so than type 1 diabetes (however, the genetics of type 2 diabetes is poorly understood).
This form encompasses → individuals who have relative (rather than absolute) insulin deficiency and have peripheral IR.
At onset, most patients with type 2 DM do not require insulin to survive, but over time their insulin secretory capacity tends to deteriorate, and many, eventually need insulin treatment to achieve optimal glucose control.
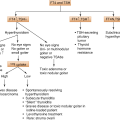
Diabetic ketoacidosis (DKA) seldom occurs spontaneously in type 2 diabetes; when seen, it usually arises in association with the stress of another illness such as infection or with the use of certain drugs (e.g., corticosteroids and atypical antipsychotics). Although the pathogenesis is not fully understood, autoimmune destruction of β cells does not occur and patients do not have any of the other known causes of diabetes. The proposed pathways involved in the pathogenesis of type 2 DM are detailed in the (Fig. 8.1).
Obesity and Insulin Resistance in Type 2 DM
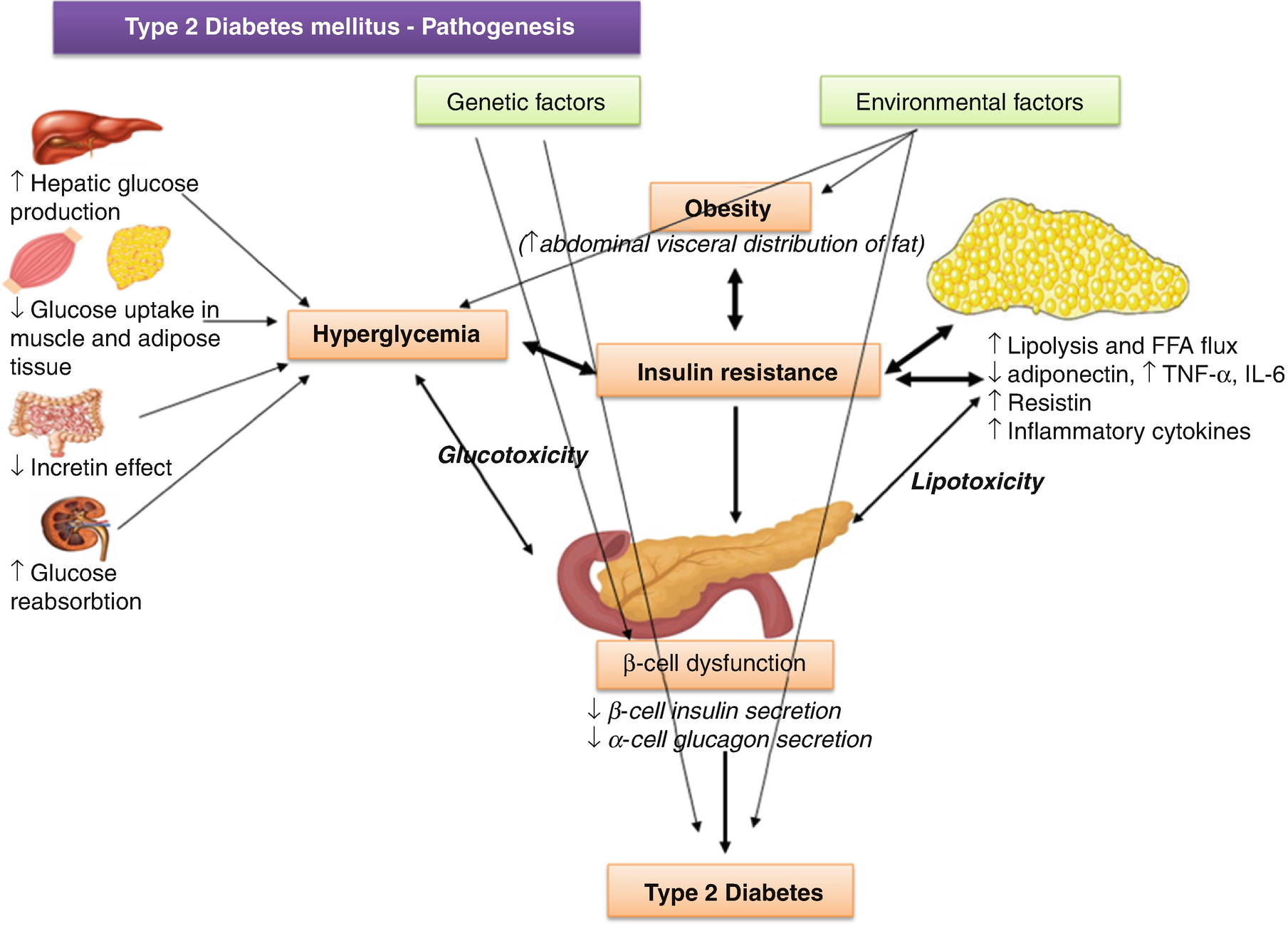
Type 2 DM pathogenesis: proposed pathways
Adipokines, Free Fatty Acids and Ectopic Lipid Storage
Adipose tissue can affect the insulin sensitivity of other tissues through the secretion of signaling molecules, adipokines, that inhibit (tumor necrosis factor α [TNF-α], interleukin [IL-6], leptin, resistin, etc.) or enhance (adiponectin) insulin signaling locally or in distal target tissues (Fig. 8.1). Levels of fat storage in adipocytes, along with insulin signaling itself, regulate the production and secretion of many of the adipokines. The release of FFAs by engorged adipocytes, especially visceral adipocytes, may play a role in the development of IR as well. Oxidation of FFAs by muscle and other tissues could inhibit glycolysis and reduce insulin-stimulated glucose removal (the Randle hypothesis). Increased fat storage in adipocytes and release of FFAs may also eventually cause a shift in lipid storage, increasing lipid uptake and storage in nonadipose tissues such as muscle, liver, and β cells. Ectopic lipid storage in these tissues may lead to a decrease in their insulin sensitivity.
Inflammation
Adipose tissue contains a variety of other cell type, besides adipocytes, including inflammatory/immune cells, such as macrophages and lymphocytes that appear to be implicated in obesity-induced IR, as well. The activated macrophages release TNF-α, IL-6, nitrix oxide, etc., that decrease the insulin sensitivity of the adipocytes and further increase their release of proinflammatory fatty acids and peptides, creating a positive feedback loop that maintains a chronic state of local inflammation and IR (Fig. 8.1). Release of these adipokines and proinflammatory cytokines, along with the increased release of FFA and the development of ectopic lipid accumulation, promotes the development of inflammation and IR in the other key insulin-target tissues, such as muscle and liver. Similar mechanisms could also lead to inflammation in the islets and contributes to β-cell failure. On the other hand, peroxisome proliferator activated receptor (PPAR)-γ, highly expressed in the adipose tissue, has beneficial effects on systemic insulin signaling.
Β-cell Defects in Type 2 DM
Although the majority of people with type 2 DM have IR, most people with IR do not have DM because their β cells compensate for the IR by producing and secreting more insulin. Those individuals who develop type 2 DM have a defect in the compensatory response of their β cells to IR. Functionally, this defect is revealed by a reduction in first phase insulin secretion and the maximal insulin secretion stimulation by glucose. While increased insulin secretion per cell may contribute to the compensatory response to IR, increases in the number of β cells play a role as well. In the setting of obesity, hyperplasia of pancreatic β cells is often present and probably accounts for the normal or exaggerated insulin response to the glucose and other stimuli seen in obese individuals without type 2 DM. Several possible defects contribute to the failure of cell mass compensation in people with type 2DM. In susceptible individuals with obesity, ectopic fat deposition in the islets, local obesity-induced inflammation in the islet and local and circulating adipokines and inflammatory cytokines may accelerate β-cell loss. As β-cell failure progresses, levels of glucose and FFA start to rise, which in turn can cause further β-cell toxicity. Once DM is established, all of these mechanisms may further contribute to the progressive decline in cell function that characterizes type 2 DM.
Genetics of Type 2 DM
So far, >70 genetic susceptibility variants for type 2 diabetes have been detected, largely through GWAS.
TCF7L2 (encoding transcription factor 7-like 2 protein involved in Wnt signaling and implicated in β-cell turnover) in chromosome 10q is the susceptibility locus, with the largest effect on type 2 diabetes risk, described to date. Inheritance of the high-risk TCF7L2 allele increases the probability of developing DM by 1.5-fold.
Among the genes identified so far, most are involved in β-cell function and turnover.
Together, the genetic loci found so far only explain a small proportion (≈10%) of the overall heritable risk for type 2 diabetes.
Ongoing research is focused on identifying rare genetic variants, which might account for the missing heritability in type 2 diabetes.
Environmental Factors in Type 2 DM
Besides genetics, environment also plays a critical role in type 2 DM: it determines the age of the onset of the disease as well as its severity. The risk of developing type 2 DM is known to increase with age, obesity, and lack of physical activity. The prevalence of this disease is higher in Western countries and westernizing countries in comparison to underdeveloped countries. The higher the rates of obesity, the more increased the incidence of type 2 DM worldwide, indicating that people have more and more access to food with high caloric intake and lack physical activity, which ↑ adiposity, especially in the more readily mobilized fat stores surrounding the viscera in the abdomen. This increased visceral adiposity also accounts for the dramatic growing incidence of type 2 DM in children.
8.2.4.3 Monogenic Diabetes Syndromes
Monogenic defects that cause β-cell dysfunction, such as neonatal diabetes and MODY, represent a small fraction of patients with diabetes (<5%).
Maturity Onset Diabetes of the Young
MODY, the commonest cause of monogenic β-cell dysfunction, is estimated to be the underlying cause in 0.5–1% of all patients with diabetes. A typical ‘MODY patient’ has young age of onset (<45 years, frequently <25 years) and has diabetes not characteristic of type 1 or type 2 as well as a family history of diabetes that occurs in successive generations. MODY is characterized by impaired insulin secretion with minimal or no defects in insulin action (in the absence of coexistent obesity) and absence of autoimmune markers. It is inherited in an autosomal dominant pattern with abnormalities in at least 13 genes on different chromosomes identified to date. The most commonly reported forms are GCK-MODY (MODY2) (← heterozygous mutations in gene encoding the enzyme glucokinase), HNF1A-MODY (MODY3) (← heterozygous mutations in gene encoding the nuclear transcription factors hepatocyte nuclear factor 1A), and HNF4A-MODY (MODY1) (← heterozygous mutations in gene encoding the hepatocyte nuclear factor 4A).
GCK-MODY (Previous Name MODY2)
Accounts for 30–70% of MODY cases.
Results in a raised threshold for glucose-stimulated insulin secretion, but importantly insulin secretion remains regulated.
Lifelong, mild, stable fasting hyperglycemia (FPG 5.5–8 mmol/L and HbA1c <8%).
Diabetes-related microvascular complications are not observed.
Patients should be managed on diet alone (insulin is usually only required during pregnancy if the fetus is macrosomic).
HNF1A-MODY (Previous Name MODY3)
Accounts for 30–70% of MODY cases.
Associated with a progressive defect of insulin secretion that can be complicated by severe hyperglycemia (one-third patients eventually require insulin therapy) and frequent microvascular complications.
Low renal threshold for glucose.
Importantly, these patients are exquisitely sensitive to sulfonylureas.
Low doses (e.g., gliclazide 40 mg od) are recommended as first-line pharmacological therapy, often maintaining excellent glycemic control for years.
HNF4A-MODY (Previous Name MODY1)
Accounts for 5–10% of MODY cases.
Clinical presentation similar to HNF1A-MODY (except normal renal glucose threshold).
Also sensitive to sulfonylureas.
Neonatal hyperinsulinemia and hypoglycemia associated with macrosomia in affected neonates.
Neonatal Diabetes
Affects 71 in 100,000–150,000 live births.
Diabetes occurring under 6 months of age is termed “neonatal” or “congenital” diabetes, and about 80–85% of cases can be found to have an underlying monogenic cause. Neonatal diabetes occurs much less often after 6 months of age, whereas autoimmune type 1 diabetes rarely occurs before 6 months of age. Neonatal diabetes can be either transient or permanent. Transient diabetes is most often due to overexpression of genes on chromosome 6q24, usually remits within 3 months, is recurrent in about half of cases, and may be treatable with medications other than insulin. Permanent neonatal diabetes is most commonly due to autosomal dominant mutations in the genes encoding the Kir6.2 subunit (KCNJ11) and SUR1 subunit (ABCC8) of the β-cell KATP channel. Correct diagnosis has critical implications because most patients with KATP-related neonatal diabetes will exhibit improved glycemic control when treated with high-dose oral sulfonylureas instead of insulin. Insulin gene mutations are the second most common cause of permanent neonatal diabetes (10–15% cases), and, while treatment presently is intensive insulin management or insulin sensitizers, there are important genetic considerations, as most of the mutations that cause diabetes are dominantly inherited.
Note: Refer those diagnosed before 6 months for genetic investigation, even if many years later and attending adult clinics → diagnosis could change management.
Mitochondrial Diabetes
Because of mutations in mitochondrial DNA (most common is an A-to-G point mutation at position 3243 in the tRNA leucine gene).
Maternal inheritance.
Commonly associated with sensorineural deafness (MIDD: maternally inherited diabetes and deafness).
Clinical features include CNS disease, ophthalmic disease (e.g., macular retinal dystrophy), myopathy, cardiac disease (e.g., left ventricular hypertrophy (LVH)), and renal disease.
Clinical phenotype can be variable, even within the same family, due to heteroplasmy.
Patients should be referred to clinical genetics team for advice.
8.2.4.4 Gestational Diabetes
Any glycemic abnormality diagnosed during pregnancy or glucose intolerance that develops or is first recognized during pregnancy
Pregnancy in Women with Preexisting Diabetes
If diabetes is poorly controlled in the first week of pregnancy, the risks of spontaneous abortion and congenital malformation (e.g., caudal regression, anencephaly, spina bifida, hydrocephalus or other CNS defects, cardiac anomalies: transposition of great vessels, ventricular septal defect atrial septal defect, anal/rectal atresia, renal anomalies, situs inversus) of the infant are increased.
Poor glycemic control later in pregnancy is associated with still births, fetal macrosomia, polyhydramnios, and neonatal hypoglycemia.
Preexisting diabetes-related complications, particularly gastroenteropathy, retinopathy, and nephropathy can impact both the pregnant woman and the fetus.
DM in pregnancy
Gestational diabetes mellitus (GDM)
- 1.
Women diagnosed with diabetes by standard diagnostic criteria in the first trimester should be classified as having preexisting pregestational diabetes (type 2 DM or, very rarely, type 1 DM, or monogenic diabetes) (Table 8.3).
- 2.
GDM is diabetes that is first diagnosed in the second or third trimester of pregnancy that is not clearly either preexisting type 1 or type 2 diabetes.
Overt DM in pregnancy-diagnosis
Measure of glycemia FPGa A1ca Random plasma glucoseb | Consensus threshold ≥7.0 mmol/L (126 mg/dL) ≥6.5% (DCCT/UKPDS standardized) ≥11.1 mmol/L (200 mg/dL) + confirmation |
GDM screening—increased risk subjects
Overweight or obesity (BMI > 25 kg/m2) pre-pregnancy Excessive weight gain during the first 6 months of pregnancy Mother older age (over 37 years) Multiparity History of IFG or IGT Members of ethnic populations at high risk for GDM Prior history of GDM or delivery of large-for-gestational-age infant Excessive weight gain History of macrosomia/polyhydramnios or fetal hypotrophy History of fetal mortality Diagnosis of PCOS Strong family history of type 2 diabetes |
The risk assessment for GDM should be performed at the first prenatal visit (Table 8.4). Those women at high risk should undergo OGTT testing as soon as feasible. If the test is negative, then they should be retested at 24–28 weeks of gestation. Lower risk women are screened for GDM using a two-step protocol performed at 24–28 weeks (not detailed here).
Diagnosis of Gestational Diabetes
Screening for and diagnosis of GDM
One step strategy (75-g OGTT): The diagnosis of GDM is made when any of the following plasma glucose values are met or exceeded: Fasting: 92 mg/dL (5.1 mmol/L) 1 h: 180 mg/dL (10.0 mmol/L) 2 h: 153 mg/dL (8.5 mmol/L) |
8.2.5 Clinical Presentation of Diabetes Mellitus
Hyperglycemia symptoms: “3 P”—polyuria —polydipsia —polyphagia Weight loss Weakness Irritability, insomnia Ketoacidosis Nausea, vomiting Complications at onset Infections, candidiasis |
Ketoacidosis Severe dehydration—dry skin A distinctive fruity odor on the breath-ketones Increased rate of breathing-acidotic breath—Kussmaul Abdominal pain Nausea, vomiting Altered state, coma |
Type 1 DM | Type 2 DM |
|---|---|
Onset: usually <30 years of age Body habitus: normal to thin Osmotic diuresis Nocturnal enuresis due to polyuria may signal the onset of diabetes in very young children Blurred vision Weight loss, despite normal or ↑ appetite Dizziness and weakness Paresthesias Ketoacidosis → anorexia, nausea, and vomiting, impaired consciousness Stupor or even coma Rapid deep breathing and the fruity breath odor of acetone | Onset: usually >40 years of age; ↑ incidence in pediatric population ← to obesity Body habitus: typically overweight with increased central obesity Chronic skin infections; generalized pruritus and symptoms of candidal vaginitis ± polyuria, thirst, blurred vision, paresthesias, and fatigue Obesity or overweight—centripetal fat distribution Acanthosis nigricans—hyperpigmented, hyperkeratotic skin in the axilla, groin, and back of neck ← significant insulin resistance Hypertension Eruptive xanthomata on the flexor surface of the limbs and on the buttocks and lipemia retinalis ← hyperchylomicronemia |
8.2.6 Diagnostic Workup—Type 1 DM
C Peptide (released in a 1:1 ratio with insulin as this is secreted)
Markers of the immune destruction of the β cell include (islet cell Ab in up to 60–95%):
islet cell autoantibodies (ICA)
autoantibodies to insulin (IAA)
autoantibodies to glutamic acid decarboxylase (GAD65) (= the most common one)
autoantibodies to the tyrosine phosphatases IA-2 and IA-2β
ZnT8 autoantibodies
Plasma glucose values—glycemic profile
fasting
postprandial (90–120 min)
nocturnal (3 a.m.)
Glycated hemoglobin A1c
Serum creatinine
Albuminuria/proteinuria
Lipid profile: total cholesterol, low-density lipoprotein (LDL)-cholesterol, high-density lipoprotein (HDL)-cholesterol, triglycerides
Full blood cell count
Thyroid function tests
Other biomarkers of autoimmune disease (celiac disease)
8.2.7 Diagnostic Workup Type 2 DM
Plasma glucose values—according to treatment
fasting
postprandial (90–120 min)
± nocturnal (3 a.m.) (if on insulin)
Glycated hemoglobin A1c
Serum creatinine
Albuminuria/proteinuria
Lipid profile: total cholesterol, LDL-cholesterol, HDL-cholesterol, TG
C Reactive Protein
Fibrinogen level
Liver enzymes: aminotransferases, gamma-GT,
Uric acid
8.2.8 Treatment of Diabetes Mellitus
Main Objectives (Table 8.6)
Prevention of chronic complications
↑ quality of life
Increase life expectancy
Key Priorities of Care
Provide individualized and ongoing culturally sensitive nutritional advice.
Set individualized target glycated hemoglobin (HbA1c) with the patient, and provide a level of care to achieve and maintain that target.
Offer self-monitoring of blood glucose as an integral part of self-management, and agree when it should be performed and how it should be interpreted and acted upon. When starting insulin therapy, employ a structured training program with active dose titration.
Therapeutic targets in DM
Glycemic control A1c < 7% (6.5%) Fasting and pre-prandial glycemia <80–130 mg/dL Postprandial glycemia <145 mg/dL (180) No hypoglycemia (<70 mg/dL) |
Blood pressure (BP) control BP < 140/80 mmHg (130/70 mmHg) (ESC/ESH guidelines 2018) |
Lipid control Cholesterol <175 mg/dL LDL < 100 mg/dL (70 mg/dL if cardiovascular disease is present) TG < 150 mg/dL HDL > 40 (men)—50 (women) mg/dL |
Weight control 5–10% weight loss (in obese patients) |
8.2.8.1 Lifestyle Optimization: Diet and Weight Control Measures
Optimal diet
Regular exercise
Smoking cessation
Moderate/no alcohol consumption
Dietary/Lifestyle Advice
- (a)
Optimal Nutrition
Caloric Intake
Basal metabolic rate: 20–25 kcal/kg/day
Caloric supplementation according to physical activity level:
Sedentary: +30% kcal
Moderate physical exercise: +50% kcal
Intense physical activity: +100% kcal
Special situations—obese patients: in order to lose weight, a 500–1000 kcal reduction can be made from the calculated energy intake.
Note: In general, for the overweight and obese patients, an initial weight loss target of 5–10% is advised while remembering that any weight loss is beneficial!
For selected patients, medical or surgical options for weight loss should be considered: orlistat, phentermine/topiramate, naltrexone/extended-release bupropion and high-dose liraglutide (3 mg/day) are weight loss medications approved in combination with diet and exercise.
Eating Patterns and Macronutrient Distribution
Carbohydrate intake from vegetables, fruits, legumes, whole grains, and dairy products, with an emphasis on foods higher in fiber and lower in glycemic load, is preferred over other sources, especially those containing added sugars.
Include low-fat dairy products and oily fish at least two times (two servings) per week!
Control the intake of foods containing saturated and trans fatty acids!
Remember that alcohol is a significant source of calories, and advise accordingly!
For people with type 1 diabetes and those with type 2 diabetes who are prescribed a flexible insulin therapy program, education on how to use CH counting and in some cases fat and protein gram estimation to determine mealtime insulin dosing is recommended to improve glycemic control.
For individuals whose daily insulin dosing is fixed, a consistent pattern of carbohydrate intake with respect to time and amount may be recommended to improve glycemic control and reduce the risk of hypoglycemia.
People with diabetes and those at risk should avoid sugar-sweetened beverages in order to control weight and reduce their risk for CVD and fatty liver and should minimize the consumption of foods with added sugar that have the capacity to displace healthier, more nutrient-dense food choices.
Data on the ideal total dietary fat content for people with diabetes are inconclusive, so an eating plan emphasizing elements of a Mediterranean-style diet rich in monounsaturated and polyunsaturated fats may be considered to improve glucose metabolism and lower CVD risk and can be an effective alternative to a diet low in total fat but relatively high in carbohydrates.
The amount of dietary saturated fat, cholesterol, and transfat recommended for people with diabetes is the same as that recommended for the general population—LOW !!
As for the general population, people with diabetes should limit sodium consumption to <2300 mg/day, although further restriction may be indicated for those with both diabetes and hypertension.
- (b)
Physical Activity—Recommendations
Children and adolescents with type 1 or type 2 diabetes or prediabetes should engage in 60 min/day or more of moderate- or vigorous-intensity aerobic activity, with vigorous muscle-strengthening and bone-strengthening activities at least 3 days/week.
Most adults with type 1 and type 2 diabetes should engage in 150 min or more of moderate- to vigorous-intensity aerobic activity per week, spread over at least 3 days/week, with no more than 2 consecutive days without activity. Shorter durations (minimum 75 min/week) of vigorous-intensity or interval training may be sufficient for younger and more physically fit individuals.
Adults with type 1 and type 2 diabetes should engage in 2–3 sessions/week of resistance exercise on nonconsecutive days.
- (c)
Metabolic Surgery
Metabolic surgery should be recommended as an option to treat type 2 diabetes in appropriate surgical candidates with BMI ≥ 40 kg/m2 (BMI ≥ 37.5 kg/m2 in Asian Americans), regardless of the level of glycemic control or complexity of glucose-lowering regimens, and in adults with BMI 35.0–39.9 kg/m2 (32.5–37.4 kg/m2 in Asian Americans) when hyperglycemia is inadequately controlled despite lifestyle and optimal medical therapy.
Metabolic surgery should be considered as an option for adults with type 2 diabetes and BMI 30.0–34.9 kg/m2 (27.5–32.4 kg/m2 in Asian Americans) if hyperglycemia is inadequately controlled despite optimal medical control by either oral or injectable medications (including insulin).
8.2.8.2 Pharmacotherapy of Diabetes
- 1.
Agents for the Treatment of Hyperglycemia (Other Than Insulin)
- (a)
Drugs That Act on Insulin Target Tissues
Biguanides
Metformin is first-line therapy for the majority of patients with type 2 DM, particularly the obese or overweight (Fig. 8.2).
Drugs/noninsulin for treatment of type 2 diabetes
Biguanides–metformin |
Sulfonylureas: Glyburide 1.25, 2.5, and 5 mg Gliclazide, Glimepiride (Amaryl®): 1, 2, 4 mg |
Meglitinide analogs: Repaglinide (Prandin®) 0.5, 1, and 2 mg tablets and d-phenylalanine derivative (Nateglinide) |
Thiazolidinediones: rosiglitazone (Avandia®), pioglitazone (Actos®) |
α-Glucosidase inhibitors: Acarbose 50 mg twice daily–100 mg three times daily |
Incretin mimetics GLP-1 receptor agonists (GLP-1RA): Exenatide (Bydureon®, Byetta®), Liraglutide (Victoza®) DPP-4 inhibitors: Sitagliptin, Vildagliptin, Saxagliptin, Linagliptin |
Inhibitors of sodium-glucose cotransporters (SGLT2 inhibitors): dapagliflozin, empagliflozin |
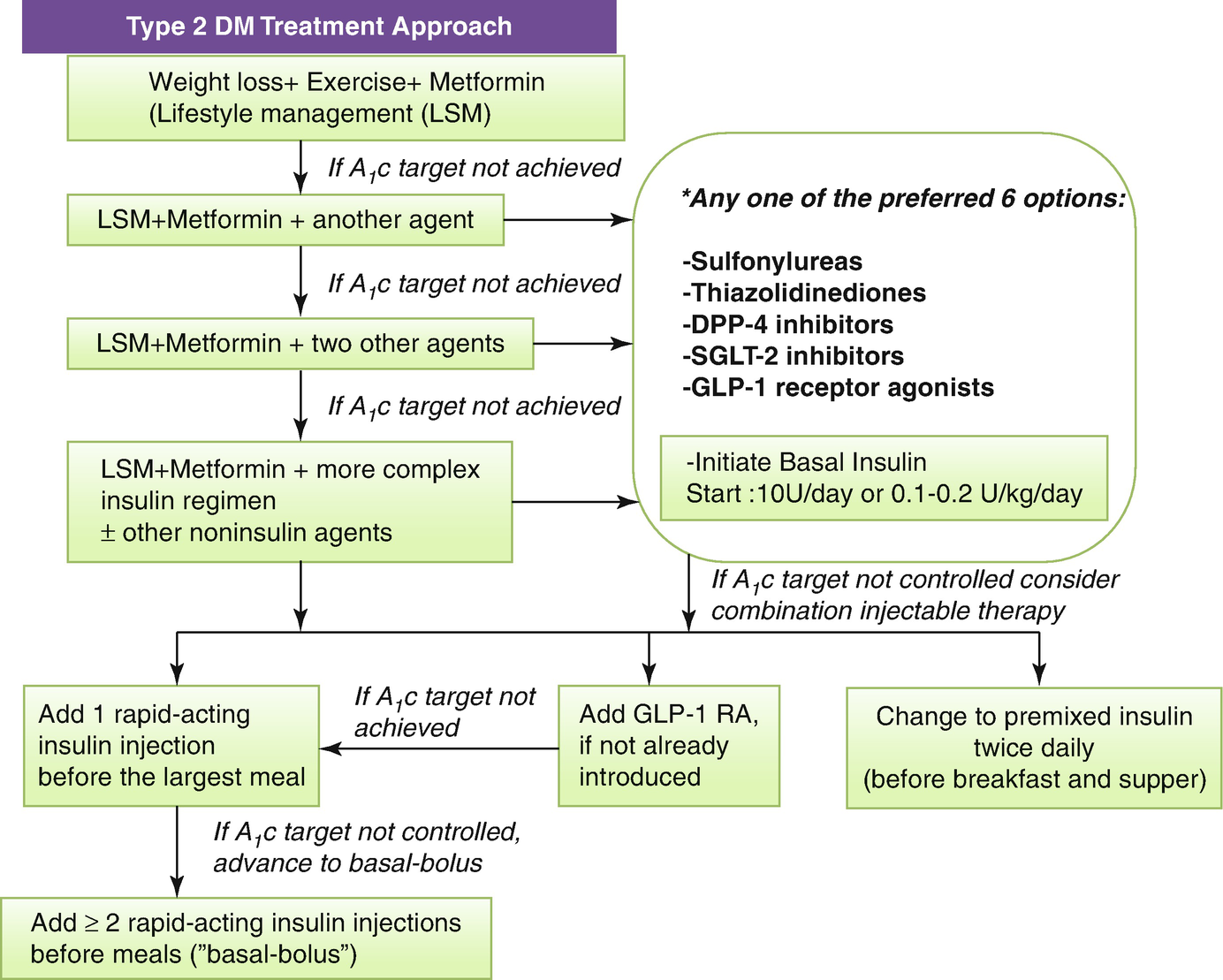
An algorithm for treating type 2 DM. *In making a therapeutic decision take into account efficacy, hypoglycemia risk; effect on weight, side effects, contraindications, cost, patient factors and associated disorders: eg ASCVD, CKD
Works (primarily in the liver) by ↓ hepatic gluconeogenesis and lipogenesis and ↑ muscle glucose uptake/metabolism.
Acts primarily through AMPK, which it activates by uncoupling mitochondrial oxidative phosphorylation and increasing cellular AMP levels.
The current recommendation is to start this drug at diagnosis, unless there are contraindications. Metformin is effective and safe and is inexpensive; it improves both fasting and postprandial hyperglycemia and hypertriglyceridemia in obese patients with diabetes without the weight gain (weight neutral) associated with insulin or sulfonylurea therapy; additionally, it may reduce risk of cardiovascular events and death.
With long-term use, a 0.8–2.0% (or 9–22 mmol/mol) reduction in HbA1c can be expected.
Start with a low dose and increase the dosage very gradually in divided doses—taken with meals—to reduce minor gastrointestinal upsets. A common schedule would be one 500 mg tablet three times a day with meals or one 850 or 1000 mg tablet twice daily at breakfast and dinner. The maximum recommended dose is 850 mg three times a day. Hypoglycemia does not occur with therapeutic doses of metformin.
Contraindications
It is contraindicated if estimated glomerular filtration rate (eGFR) is <30 mL/min/1.72 m2.
Likewise, patients with liver failure or cardiac impairment or abusers of ethanol should not receive this drug.
Side Effects
Thiazolidinediones (glitazones)—Peroxisome Proliferator–Activated Receptor Agonists
Mode of Action
Insulin-sensitizing agents by activating the peroxisome proliferator activated receptor (PPAR-γ) which stimulates gene transcription for glucose transporter molecules, such as Glut 1 and Glut 4.
Other observed effects of thiazolidinediones include ↓ FFA levels; ↓ hepatic glucose output; ↑ adiponectin and ↓ resistin release from adipocytes; and ↑ differentiation of preadipocytes into adipocytes. They have also been demonstrated to ↓ levels of plasminogen activator inhibitor type 1, matrix metalloproteinase 9, C-reactive protein, and interleukin-6 (IL-6). Like the biguanides, this class of drugs does not cause hypoglycemia.
Preparations
Rosiglitazone: 4–8 mg daily.
Pioglitazone: 15–45 mg daily.
Both are effective as monotherapy and in combination with sulfonylureas (SU), metformin, or insulin (Fig. 8.2).
As monotherapy: ↓ HbA1c by about 1–2% points.
Side Effects
Patients should be warned about the possibility of significant edema and weight gain. Pioglitazone should not be used in people with evidence of heart failure and those at a higher risk of bone fracture and osteoporosis.
- (b)
Insulin Secretagogues
Sulfonylureas
Mode of Action
Sulfonylureas (SU) stimulate β-cell potassium ATP channel, closing the potassium channel that leads to membrane depolarization, calcium influx, and subsequent insulin release. A doubling of glucose-stimulated insulin secretion can be expected.
Results in a 1–2% (or 11–22 mmol/mol) reduction in HbA1c long term.
Consider as first line in nonobese patients, those unable to tolerate metformin, or if a rapid response to therapy is required because of hyperglycemic symptoms. Can be used as second-line agents when blood glucose control is inadequate with metformin.
Side effects and Contraindications
Predominantly hypoglycemia and weight gain.
SU are generally contraindicated in patients with severe liver or kidney impairment.
SU are not indicated for use in patients with type 1 diabetes since these drugs require functioning pancreatic β cells to produce their effect on blood glucose.
First-generation sulfonylureas (tolbutamide, tolazamide, acetohexamide, and chlorpropamide): rarely used.
Second-generation sulfonylureas: glyburide, glipizide, gliclazide, and glimepiride—these agents are highly potent (100- to 200-fold more so than tolbutamide). The drugs should be used with caution in patients with CVD as well as in elderly patients, in whom hypoglycemia would be especially dangerous.
Glyburide (glibenclamide)—the usual starting dose is 2.5 mg/day, and the average maintenance dose is 5–10 mg/day given as a single morning dose.
Gliclazide (not available in the United States)—the recommended starting dose is 40–80 mg/day with a maximum dose of 320 mg (as divided doses before breakfast and dinner). The drug is metabolized by the liver.
Meglitinide Analogs (Repaglinide) and d-Phenylalanine Derivative (Nateglinide)
They also act by binding to the sulfonylurea receptor and closing the ATP-sensitive potassium channel and stimulate insulin secretion. Like the SU, repaglinide can be used in combination with metformin.
Hypoglycemia and weight gain are the main side effects.
Dose: 0.5 mg three times a day 15 min—up to 16 mg, before each meal.
- (c)
Drugs That Affect Glucose Absorption
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree