Endocrine Neoplasms
Harold E. Carlson
I. GENERAL CONSIDERATIONS.
Cancers of endocrine glands constitute about 3% of all malignancies. Most malignant neoplasms derived from endocrine organs are not associated with clinical endocrinopathies, although several do produce unique syndromes and biochemical markers.
A. Steroid hormones are usually produced by the tissue that normally produces them, such as the adrenal cortex and gonads, whether that tissue is healthy or cancerous. Occasionally, human chorionic gonadotropin (hCG)-producing tumors of the placenta or other organs (e.g., lung) have the capacity to transform androgens into estrogens. The mechanism of action for most steroid hormones depends on specific receptors in the target cell cytoplasm or nucleus.
B. Peptide hormones and catecholamines appear to act at the cell surface, where they attach to specific receptors and modify intracellular concentrations of cyclic nucleotides, calcium, and kinases.
1. Amine precursor uptake and decarboxylation (APUD) cells are theoretically derived from embryonic neuroectoderm (melanocytes, thyroid C cells, adrenal medulla, paraspinal ganglia, argentaffin cells of the intestine). These cells produce hormone mediators such as serotonin, catecholamines, histamine, and kinins. Neoplasia of these tissues gives rise to carcinoid tumors, pheochromocytoma, and medullary thyroid cancer; these tumors may also produce peptide hormones (e.g., adrenocorticotropic hormone [ACTH] and vasoactive intestinal polypeptide [VIP]) in addition to their natural products. Other peptide-producing endocrine tissues (e.g., parathyroid, pancreatic islet) demonstrate some APUD characteristics, even though they may not be derived from neuroectoderm.
2. Peptide hormones, such as ACTH, hCG, and calcitonin, are produced by a wide variety of neoplastic tissues that may or may not normally synthesize detectable amounts of these hormones. Many of these peptides are synthesized as a pre’hormone. A segment of prehormone is enzymatically cleaved to form a storage molecule, a prohormone. The prohormone is further cleaved into the active hormone that is secreted into the blood.
3. Gastrointestinal hormones, such as insulin, glucagon, somatostatin, VIP, and gastrin, are normally produced by gut endocrine cells and the pancreatic islets. Neoplasms of these tissues commonly produce one or more of these hormones; gut hormones are also normally produced in the brain and may be products of a wide variety of other neoplasms.
C. Multiple endocrine neoplasias (MEN) are inherited, mendelian-dominant, endocrine tumor syndromes. Two categories of the syndrome are recognized.
1. MEN-1 (Wermer syndrome; menin tumor-suppressor gene located at chromosome 11q13)
a. Pituitary tumors (acromegaly, nonfunctioning adenoma, prolactinoma, or ACTH-producing adenoma)
b. Pancreatic islet cell tumors, including gastrinoma, VIPoma, glucagonoma, and insulinoma
c. Parathyroid adenomas
2. MEN-2. Medullary carcinoma of the thyroid is present in all patients with this syndrome. Cushing syndrome may develop as a consequence of ectopic ACTH production by medullary carcinoma or pheochromocytoma.
a. MEN-2A (Sipple syndrome; ret oncogene located at chromosome 10q11)
(1) Medullary carcinoma of the thyroid
(2) Pheochromocytoma (bilateral)
(3) Parathyroid hyperplasia or adenomas
b. MEN-2B (ret oncogene located at 10q11)
(1) Medullary carcinoma of the thyroid
(2) Pheochromocytoma (bilateral)
(3) Multiple mucosal ganglioneuromas (lips, tongue, eyelids)
(4) Marfanoid body habitus, high-arched palate, pes cavus, diverticulae, and sugar-loaf skull often accompany the endocrine abnormalities in MEN-2B.
II. CARCINOID TUMORS
A. Epidemiology and etiology. Carcinoid cancers represent <1% of visceral malignancies. The cause of these tumors is unknown, but they are sometimes associated with MEN-1.
B. Pathology and natural history
1. Primary tumor. Carcinoid tumors belong to the APUD system of tumors (see Section I.B.1). The primary tumors are usually small and most commonly arise in the small intestine. They may also develop in the stomach, colorectum, lung, ovary, and rarely other organs. Appendiceal carcinoids are common but are usually of no clinical significance.
2. Metastases tend to develop primarily in the liver. Bone metastases, which are often osteoblastic, also occur. Carcinoid metastases are indolent or slowly progressive and evolve over many years. Carcinoid tumors tend to produce desmoplastic responses, which can result in mesenteric fibrosis and bowel obstruction (“parachute intestine”). Hormonally inactive tumors usually cause death by replacing hepatic tissue, which leads to liver failure.
3. Tumor products. Hormonally active tumors occur in 30% to 50% of patients and produce a variety of potentially lethal complications (carcinoid syndrome).
a. Small intestine carcinoids never produce the carcinoid syndrome in the absence of liver metastases; the responsible hormonal mediators are degraded in their first pass through the liver.
b. Benign and malignant lung carcinoids occur with about equal frequency; those that produce the carcinoid syndrome are malignant. Lung carcinoids can potentially produce hormonal effects without metastasizing; active tumor products may pass directly into the circulation without being filtered by the liver. Most patients with endocrinologically active lung carcinoids, however, also have liver metastases. Bronchial carcinoids that produce ACTH or growth hormone-releasing hormone (GH-RH) may be benign, and Cushing syndrome or acromegaly may be the only endocrine manifestation.
c. Symptomatic ovarian carcinoids are rarely associated with liver metastases.
d. Humoral mediators of the carcinoid syndrome are serotonin, histamine, kinins, prostaglandins, and other hormonally active tumor products.
(1) The major source of serotonin is dietary tryptophan, which normally is mostly metabolized to nicotinic acid. In carcinoid syndrome, tryptophan metabolism is directed to the production of serotonin (Fig. 15.1). Most patients with carcinoid syndrome develop chemical
evidence of niacin deficiency, and some may develop clinically recognizable pellagra.
evidence of niacin deficiency, and some may develop clinically recognizable pellagra.
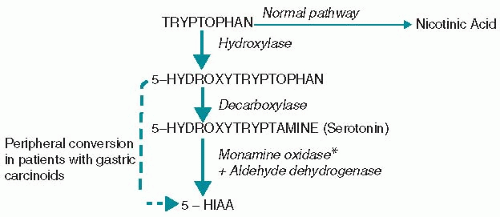 Figure 15.1. Hepatic metabolism of tryptophan and serotonin in carcinoid syndrome. The normal pathway (thin arrow) of tryptophan metabolism is impaired in carcinoid syndrome, resulting in excessive production of serotonin. *Monamine oxidase inhibitors interfere with the metabolism of serotonin and are contraindicated in patients with carcinoid syndrome. 5-HIAA, 5-hydroxyindoleacetic acid. |
(2) Other hormones and hormone metabolites that are found in some patients with carcinoid include calcitonin, gastrin, GH-RH, and ACTH. These substances may or may not produce clinical syndromes, but they should be searched for in patients with carcinoid and serum calcium abnormalities, peptic ulcer, acromegaly, or Cushing syndrome.
C. Diagnosis
1. Symptoms: Endocrinologically inactive carcinoids. Most carcinoid tumors are endocrinologically inactive. Patients who have these tumors may have appendicitis, bowel obstruction, or a painful, enlarged liver that results from metastases. Bronchial carcinoids may produce cough, hemoptysis, or frequent pulmonary infections.
2. Symptoms: Endocrinologically active carcinoids
a. Humoral mediators produce attacks of flushing, diarrhea, hypotension, light-headedness, and bronchospasm in various combinations. Attacks may be spontaneous or precipitated by emotional stress, alcohol ingestion, exercise, eating, or vigorous palpation of a liver that contains metastatic deposits.
b. Heart failure from valvular lesions commonly occurs in patients with long-standing carcinoid symptoms and appears to be related to serotonin excess. Ileal carcinoids with hepatic metastases produce tricuspid and/or pulmonic valve stenosis and insufficiency. A patent foramen ovale or bronchial carcinoids with venous drainage into the left atrium can occasionally produce mitral valve disease.
3. Physical findings
a. The characteristic flush differs somewhat according to the site of the primary tumor.
(1) Ileal carcinoid. Purple flush involves the upper trunk and face and usually lasts <30 minutes.
(2) Bronchial carcinoid. Deep, dusky purple flush over the entire body
(3) Gastric carcinoid. Generalized urticaria-like, pruritic, and painful wheals, probably related to histamine production
b. Chronic skin changes may result from repeated episodes of flushing, especially with bronchial carcinoids, which cause thickening of the facial features, telangiectasis, enlargement of the salivary glands, and leonine facies. A pellagrous skin rash characterized by photosensitivity, atrophy of the lingual mucosa, and thickened skin may develop.
c. Right heart failure with evidence of tricuspid or pulmonic valve disease
d. Hepatomegaly
e. Cushing syndrome and, occasionally, acromegaly
4. Laboratory studies in all patients
a. Routine blood tests, particularly liver function tests (LFTs)
b. Liver MRI scan
c. Chest CT scan to search for bronchial carcinoids
d. Upper gastrointestinal barium series or endoscopy
e. Nuclear scanning using a radiolabeled somatostatin analog
f. A histologic diagnosis is essential for management. Biopsy the site that is associated with the least morbidity and that has been determined by noninvasive tests to be probably affected.
5. Laboratory studies in patients with symptoms have traditionally consisted of 24-hour urine collections for 5-hydroxyindoleacetic acid (5-HIAA), particularly in patients with midgut carcinoids. Serotonin is a product of tryptophan metabolism and is metabolized to 5-HIAA (Fig. 15.1). Fasting plasma 5-HIAA has been shown to be at least as sensitive and specific as urinary 5-HIAA. Platelet or urine serotonin measurements may also be useful, particularly in foregut carcinoids, in which 5-HIAA production may be minimal.
a. Causes of elevated urine or plasma 5-HIAA include the following:
(1) Carcinoid syndrome
(2) Other tumors that produce 5-HIAA include biliary, pancreatic islet, and medullary thyroid cancers.
(3) Dietary intake of nuts, bananas, avocados, or pineapples within 48 hours of urine collection or 8 hours of plasma sampling
(4) Medications that must be stopped 1 day before 5-HIAA measurement include mephenesin and guaifenesin.
(5) Malabsorption syndromes (celiac disease, Whipple disease, and tropical sprue) rarely increase 5-HIAA urine excretion above 20 mg per 24 hours.
b. Causes of falsely low 5-HIAA excretion. Phenothiazines interfere with the color reaction of the urine test and must be stopped 2 to 3 days before the collection of urine.
c. Interpretation. A urine level of 5-HIAA >9 mg per 24 hours in patients without malabsorption or >30 mg per 24 hours in patients with malabsorption is pathognomonic for carcinoid unless interfering foods or drugs have been ingested. The magnitude of 5-HIAA excretion in the urine roughly corresponds to the tumor volume; 5-HIAA excretion can also be used to monitor therapy. Normal values for plasma 5-HIAA are laboratory-dependent.
d. Chromogranin A (CgA) is a soluble protein found in secretory granules in a variety of neuroendocrine cell types. Plasma CgA is elevated in nearly all patients with carcinoid tumors, but is nonspecific, since it is also elevated in patients with other neuroendocrine tumors such as pancreatic islet tumors, small cell lung cancer, medullary thyroid cancer, and pheochromocytoma. Serum CgA may also be elevated in patients receiving proton pump inhibitors.
D. Management. The most important principle of management of metastatic carcinoid tumors is therapeutic restraint. These patients often survive for >10 years without antitumor treatment. Patients with endocrinologically active tumors are at especially high risk for complications from any procedure requiring anesthesia. Therapy should be focused on controlling the endocrine symptoms.
1. Surgery is useful for patients with localized primary carcinoids or metastatic tumors that produce obstruction. For patients with incidental appendiceal carcinoids that are ≤2 cm in diameter (which rarely metastasize), appendectomy is adequate treatment.
Partial hepatectomy has been recommended by some physicians, particularly if the metastases are confined to one lobe of the liver. The mortality rate of hepatectomy and the long natural history of the disease, however, often dissuade the physician from recommending the procedure. Palliation of liver metastases may also be accomplished with cryosurgery or radiofrequency ablation.
2. Hepatic artery occlusion performed surgically or by catheterization and embolization of hepatic metastases has been successfully used to palliate endocrine symptoms or pain. Objective regression of manifestations occurs in 60% of patients for a median of 4 months. Side effects of arterial occlusion include fever, nausea, and LFT abnormalities. Both the response rate and the median duration of response appear to improve when occlusion is followed by sequenced chemotherapy (see Section II.D.4).
3. Radiation therapy (RT) is used to palliate liver or bone pain caused by faradvanced metastatic disease unresponsive to other treatments. However, carcinoid tumors are relatively radioresistant.
4. Chemotherapy is used late in the course of disease for treatment of symptomatic metastases and for patients with severe endocrine symptoms that do not respond satisfactorily to pharmacologic maneuvers (see Section II.D.5). There is no general agreement on when (or even if) chemotherapy should be started in patients with malignant carcinoid. Single-agent therapy with 5-fluorouracil (5-FU), streptozocin, cyclophosphamide, doxorubicin (Adriamycin), dacarbazine, temozolomide, or interferon-α (IFN-α) has been associated with response rates of about 25%, with variable median durations of response. Endocrine symptoms may be palliated, but the effect of chemotherapy on survival is not known.
a. Combination chemotherapy regimens (such as streptozocin and doxorubicin) have not clearly had a more beneficial effect compared with single agents. Cisplatin in combination with etoposide is useful for anaplastic forms of neuroendocrine carcinomas. Combinations of IFN-α (3 to 10 million units three to seven times weekly) and octreotide have not been consistently more effective than monotherapy with either agent alone; a flu-like syndrome may be problematic in patients on long-term IFN treatment, and autoimmune thyroid disease may develop in some patients.
b. Hepatic arterial occlusion or embolization, with or without chemotherapy, has been performed for symptomatic hepatic metastases from carcinoid tumors or islet cell carcinomas. Substantial or complete relief from the endocrine syndromes is achieved in about 80% of selected patients, with a median duration of 18 months.
5. Pharmacologic management. It is probably not possible to control the symptoms of carcinoid syndrome completely with aggressive dietary tryptophan restriction and high-dose antiserotonin drugs alone.
a. Somatostatin analogs such as octreotide and lanreotide reduce the production of 5-HIAA and ameliorate symptoms in about 90% of patients. The drugs have a tumoristatic effect as well and prolong the time to tumor progression. The octreotide dosage is usually 100 to 600 mcg SC daily in two to four divided doses. Long-acting depot forms of octreotide and lanreotide are also available; octreotide LAR 10 to 30 mg IM or lanreotide autogel 60 to 120 mg SC are given every 28 days. Side effects of both octreotide and lanreotide include abdominal cramping, cholelithiasis, and hyperglycemia.
b. Hypotension, the most life-threatening complication of carcinoid syndrome, is mediated by kinins (and perhaps prostaglandins) and can be precipitated by catecholamines. β-Adrenergic drugs (e.g., dopamine, epinephrine) must be strictly avoided because they may aggravate hypotension. Pure α-adrenergic (methoxamine, norepinephrine) and vasoconstrictive (angiotensin) agents are preferred for treating hypotension in carcinoid syndrome.
(1) Methoxamine (Vasoxyl) is given IM at a dose of 0.5 mL (10 mg) or IV at a dose of 0.25 mL (5 mg) over 1 to 2 minutes (using a tuberculin syringe). The dose is repeated as necessary to maintain the blood pressure.
(2) Angiotensin amide (Hypertensin), rather than methoxamine, is recommended by some anesthesiologists.
(3) Corticosteroids may prevent episodes of hypotension.
c. Flushing is mediated by kinins and histamine and may respond to several agents, including the following:
(1) Prochlorperazine (Compazine), 10 mg PO four times daily
(2) Phenoxybenzamine (Dibenzyline), 10 to 20 mg PO twice daily
(3) Cyproheptadine (Periactin), 4 to 6 mg PO four times daily
(4) Prednisone, 20 to 40 mg PO daily, is useful for flushing as a result of bronchial carcinoids and occasionally for patients with other kinds of carcinoids.
(5) The combined use of H1– and H2-receptor antagonists has been effective in patients with carcinoid flush and documented hypersecretion of histamine. Diphenhydramine hydrochloride (Benadryl), 50 mg PO four times daily, plus cimetidine (Tagamet), 300 mg PO four times daily, have been used with success in some patients.
(6) Methyldopa (Aldomet) is useful in some patients.
(7) Monoamine oxidase inhibitors are contraindicated in carcinoid syndrome because they block serotonin catabolism and can aggravate symptoms (Fig. 15.1).
d. Bronchospasm is mediated by histamine and managed with aminophylline. Adrenergic agents, such as albuterol, do not appear to worsen bronchospasm for carcinoid and may also be used with caution, since they may cause hypotension.
e. Diarrhea is mediated by serotonin and is often difficult to control. A recommended sequence for treatment before the use of octreotide is as follows:
(1) Belladonna alkaloids and phenobarbital combination (Donnagel-PG), 15 mL every 3 hours as needed
(2) Loperamide (Imodium) or diphenoxylate and atropine (Lomotil) as needed
(3) Cyproheptadine (Periactin), 4 to 6 mg PO four times daily
(4) Methysergide maleate (Sansert), started at 8 to 12 mg/d and gradually increased to 20 to 22 mg/d if needed
(5) Ondansetron, 8 mg PO three times daily
f. Preparation for anesthesia*. Patients with carcinoid syndrome are at high risk for the development of flushing, bronchospasm, and hypotension (carcinoid crisis) during surgery. Stimulation of adrenergic hormone release and use of drugs that induce hypotension (morphine, succinylcholine, and curare) must be minimized.
(1) Preoperative period. Patients should be given octreotide 100 mcg SC three times daily for 2 weeks prior to surgery to block the release of tumor products.
(2) During and after surgery. Octreotide should be given intravenously at a rate of 50 mcg/h, starting before anesthesia. Increased doses may be given if flushing or hypotension occurs. Octreotide should be tapered gradually over 1 week postoperatively.
E. Special clinical problems associated with carcinoid syndrome
1. Bowel obstruction may result from dense fibrosis of the mesentery. Surgical relief is often impossible. Patients may improve with simple nasogastric decompression and fluid replacement.
2. Right ventricular failure results from tricuspid and pulmonic valve lesions. These changes develop with far-advanced carcinoid syndrome, which has a poor prognosis independent of the heart lesions. Because of the high surgical risk in these patients, valve replacement may not be warranted. Heart failure should be medically managed with diuretics.
3. Pellagrous skin lesions may be treated with daily oral vitamin preparations containing 1 to 2 mg of niacin.
III. THYROID CANCER
A. Epidemiology and etiology
1. Incidence. Thyroid cancer accounts for about 3% of visceral malignancies; there are 45,000 new cases and 1,700 cancer deaths in the United States annually. The risk increases with age. Women are affected more than men in a ratio of 3:1.
2. Radiation exposure. Radiation fallout and RT given to the neck region in intermediate doses (<2,000 cGy) for benign conditions (such as acne in teenagers, or enlarged tonsils or thymus glands in children) increase the risk for thyroid cancer, particularly the papillary type.
a. The lag time between radiation exposure and the onset of thyroid cancer averages 25 years but ranges from 5 to 50 years. Many patients younger than 20 years of age with thyroid cancer have a history of neck irradiation.
b. About 4% of patients with thyroid cancer have a history of radiation to the neck. Between 5% and 10% of patients who have a history of neck irradiation develop thyroid cancer; 25% have an abnormal thyroid by palpation.
c. Thyroid cancers after neck irradiation are often multifocal but have an indolent course and a prognosis similar to that of spontaneous tumors.
d. Neck irradiation also increases the risk for hyperparathyroidism and parotid gland tumors.
3. Hereditary factors. Medullary cancer of the thyroid may arise sporadically or as a dominantly inherited syndrome of MEN-2 (see Section I.C.2). Thyroid tumors (including papillary and follicular carcinomas), as well as breast neoplasms, also occur frequently in Cowden multiple hamartoma syndrome and in familial adenomatous polyposis (including Gardner syndrome). Several oncogenes and tumor-suppressor genes have been implicated in the pathogenesis of thyroid neoplasms.
4. Thyroid-stimulating hormone (TSH). An increased risk for thyroid cancer may be present in patients with chronic TSH elevation, such as patients with congenital defects in thyroid hormone formation.
B. Pathology and natural history. The more aggressive histologic subtypes of thyroid cancer tend to affect older patients.
1. Papillary cancers (80% of thyroid cancers in adults) affect younger patients. Histologically, the tumor cells may be arranged in either papillary or follicular patterns; the diagnosis of papillary carcinoma is based on nuclear features, not on the presence or absence of follicles. Psammoma bodies may be present in histologic sections in about 40% of these tumors. Regional lymph nodes that drain the thyroid are involved in half of patients. Distant metastases to lungs, bone, skin, and other organs occur late, if at all.
2. Follicular cancers (10% of thyroid cancers) have a peak incidence at 40 to 50 years of age. They tend to invade blood vessels and to metastasize hematogenously to visceral sites, particularly bone. Lymph node metastases are relatively rare, especially compared with papillary cancers.
3. Anaplastic cancers (1% to 2% of thyroid cancers) occur most often in patients older than 60 years of age. Anaplastic thyroid cancers are aggressive cancers, which rapidly invade surrounding local tissues and metastasize to distant organs.
4. Medullary thyroid cancers (5% to 10% of thyroid cancers) secrete calcitonin and carcinoembryonic antigen (CEA). ACTH, histaminase, and an unidentified substance that produces diarrhea may also be secreted by these tumors. Amyloid may be seen on histologic examination and is composed of calcitonin arranged in fibrils. Metastases are mostly found in the neck and mediastinal lymph nodes and may calcify. Widespread visceral metastases occur late.
5. Hürthle cell cancer is a variant of follicular carcinoma and has a relatively aggressive metastatic course.
6. Other tumors found in the thyroid include lymphomas (1% to 2% of all thyroid cancers), a variety of soft tissue sarcomas, and metastatic cancers from kidney, colon and other primary sites. Small cell cancers of the thyroid are actually lymphomas in most cases.
C. Diagnosis
1. Symptoms and signs
a. Symptoms. Some patients with thyroid cancer complain of an enlarging mass in the neck. Hoarseness may be the result of recurrent laryngeal nerve paralysis. Neck pain or dysphagia occasionally is a complaint. Patients without symptoms may have thyroid cancer discovered at thyroidectomy done for other reasons or as an incidental finding in the course of radiologic examinations of the neck (see Section III.C.3.a).
b. Physical findings. Thyroid cancer may be found on routine physical examination as a mass in the thyroid or in the midline up to the base of the tongue (thyroglossal duct remnant). Thyroid masses <1 to 2 cm in diameter often are not palpable. Patients with thyroid cancer may have a single palpable nodule; others have a normal, multinodular, or diffusely enlarged thyroid gland. Cervical lymph nodes are sometimes palpable. Anaplastic cancer is often manifested by obvious masses infiltrating the skin and soft tissues of the neck or by respiratory distress.
2. Laboratory studies
a. Routine studies. Chest radiographs and serum alkaline phosphatase levels may be obtained to look for evidence of metastatic disease in the lung, liver, or bone. Liver and bone scans and selected skeletal radiographs are indicated when the alkaline phosphatase level is elevated.
b. Thyroid scans may be obtained in nonpregnant patients with palpable abnormalities of the thyroid who have a suppressed serum TSH, in order to document the existence of a “hot” nodule. Nonfunctional “cold” nodules are found in 90% of patients with palpable nodules, both benign and malignant, but only about 10% of cold nodules prove to be cancer. Routine isotope scanning of all thyroid nodules is therefore not indicated unless serum TSH is low.
c. Thyroid ultrasonography is useful in determining the size and location of a nodule, diagnosing cystic lesions, detecting nonpalpable nodules or lymphadenopathy, and documenting the presence of features suggestive of malignancy (e.g., microcalcifications within the nodule, irregular borders, increased internal vascularity). Purely cystic lesions, found in about 10% of patients with palpable nodules, are reported to be malignant in <1% of cases. Benign and malignant lesions cannot be confidently distinguished by ultrasonography if they contain mixed solid and cystic components or are entirely solid.
d. Serum calcitonin assay. Patients with a family history of medullary thyroid cancer or other features of MEN-2 should have serum calcitonin measured. Normal basal values may require a calcitonin stimulation test using pentagastrin or calcium infusion. Patients with elevated serum calcitonin require neck exploration regardless of findings on physical examination or sonography.
3. Thyroid gland biopsy
a. Needle aspiration biopsy is invaluable for cytologic diagnosis of thyroid nodules and for preventing unnecessary thyroidectomies. Many authorities recommend needle biopsy as the first step in the evaluation of any thyroid lump. The accuracy of needle biopsy of the thyroid is >90% for benign lesions; the false-negative rate is 5% to 10%. Only about 10% of nodules are cancerous. Roughly, if 100 patients with nodules underwent needle biopsy rather than immediate thyroidectomy, and if patients with clearly benign histopathology were excluded from surgery, 1 cancer would be missed, 9 cancers would be appropriately resected, and 10 patients with benign lesions would have undergone unnecessary surgery. Therefore, the needle biopsy saves 80 of 100 patients from unnecessary surgery at the expense of missing one cancer, which is usually indolent and can be detected later. Patients with nonpalpable nodules >1.5 cm in diameter found on radiologic examinations should generally undergo sonogram-guided needle aspiration biopsy, as should those patients with smaller nodules that appear suspicious for malignancy on sonography.
b. Open biopsy. Nodules interpreted as suspicious on needle biopsy should be removed.
D. Survival and prognostic factors
1. Papillary adenocarcinomas.




Related posts:


Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
