Incidence: 240% ↑ in incidence over prior 3 decades, in part due to ↑ detection of small papillary CA; incidence of anaplastic thyroid CA declining
Subtypes: 80-85% of malignant epithelial thyroid tumors in developed countries are papillary; 3-12% medullary; 1-3% anaplastic
| ||||||||
(rearrangement in 20% of adult sporadic papillary carcinomas), BRAF, or RAS in 70% of papillary carcinomas; rarely overlap in same tumor
PAX8-PPAR in 35% follicular carcinomas, some Hurthle cell
Commonly p/w incidental solitary thyroid nodules: Median tumor size 2-3 cm; 5-10% malignant; higher percentage if radiation exposure; majority hypofunctional; presence of microcalcifications, irregular margins, spotty intranodular flow, hypervascularity are suggestive of malignancy
FNA: Accuracy of dx 70-97%; varies w/sample quality, cytopathologist skill; ˜70% benign, 4% malignant, 10% suspicious/indeterminate; 17% insufficient sample
Medullary: Familial often detected by screening w/stimulation tests/molecular analysis; sporadic by asx thyroid mass; secretory diarrhea if bulky disease w/high calcitonin
Anaplastic: Prior or concurrent dx of well-differentiated thyroid CA or benign nodular thyroid disease; rapidly ↑ palpable neck mass (median tumor size 8-9 cm); invasion into airways & recurrent laryngeal nerve leads to obstructive sx, hemoptysis, dysphagia, hoarseness; 20-50% have distant mets at dx in lung > bone, liver
Familial tumors tend to be more aggressive than nonfamilial
Prognosis: Papillary: 90-95% long-term survival; follicular: 70-80% long-term survival; distant mets strong negative prognostic indicator; prognosis ⇔ stage; anaplastic: Median survival 4-5 mos from dx
Poor prognostic factors for well-differentiated thyroid CA: Age >45 y, male sex, poorly differentiated histology, tumor size, extrathyroid extension at dx; nodal involvement does not confer ↓ survival in younger pts
Mayo Clinic Model: AGES (age, tumor grade, tumor extent, tumor size)
Papillary/follicular, under age 45: Stage I: M0; Stage II: M1
Papillary/follicular, 45+: Stage I: T1 (<2 cm confined to thyroid); Stage II: T2 (>2 but <4 cm confined to thyroid); Stage III: T3 & early nodal involvement; Stage IV: All else including M1
Medullary: Stage I: T1, node negative; Stage II: T2-T3, node negative; Stage III: T1-T3, early nodal involvement (N1a); Stage IV: All T4, N1b, M1 disease
Anaplastic: Stage IV
Cell Derivation: Papillary, follicular, Hurthle cell, anaplastic arise from follicular cells that produce thyroid hormones; Tumors usu PAX8 & TTF1 positive
| ||||||||||||
Surgery: Mainstay of tx for all subtypes, but rarely possible w/anaplastic; complications of total thyroidectomy include recurrent laryngeal nerve injury & hypocalcemia 2/2 hypoparathyroidism
Differentiated thyroid CA: Levothyroxine suppression of TSH as it is a potential growth factor for microscopic CA deposits, RAI administration to ablate any nl thyroid remnant
Met disease: RAI, external-beam radiotherapy, & chemotherapy (doxorubicin + platinum) for met disease; TKIs (eg, sorafenib) have activity in iodinerefractory disease (J Clin Oncol 2008;26:4714) & are the preferred tx by guidelines
MTC: Total thyroidectomy w/b/l central compartment node dissection & at least unilateral neck dissection; radiation & chemotherapy not considered effective; targeted therapies vandetanib (J Clin Oncol 2012;30:134) & cabozantinib (J Clin Oncol 2011;29:2660) approved for tx of met disease; treat local recurrences surgically; screen for germline RET gene Mt as total thyroidectomy is preventive in young, at-risk family members
Anaplastic thyroid CA: May require urgent tracheostomy; if unresectable at dx, radiation-based Rx, often w/chemotherapy for sensitization; RAI not useful
Presentation
Neuro sx: HA (expansion of sella), diplopia (oculomotor nerve compression), pituitary apoplexy (sudden hemorrhage into the mass), CSF rhinorrhea (inferior extension of mass), visual field deficits
Hormonal abnormalities (hyper- or hyposecretion; see below)
Incidental finding on MRI done for other reason (“incidentaloma”)
Causes
Pituitary adenomas = most common cause; ˜85% of sellar masses
Also: Physiologic pituitary enlargement (pregnancy, 1° hypothyroidism, 1° hypogonadism); cyst, abscess or AV fistula of cavernous sinus; hypophysitis (lymphocytic = most common kind, seen in postpartum ♀ or in anti-CTLA-4 tx of malignancies); benign tumors (craniopharyngioma, meningioma); 1° malignant tumors (germ cell, chordoma, 1° CNS lymphoma, sarcoma, pituitary carcinoma-rare); met disease (breast/lung 1° most common)
In a large registry of pituitary tumors (N = 4122), 84.6% were adenomas, 3.2% were craniopharyngioma, 1.8% were Rathke cysts, 1.8% were Crooke cells (w/o adenoma), ˜1% were meningiomas, 0.6% were mets, & 0.5% were chordomas. Sellar tumors of all other types each occurred no more often <0.5% of the time (CNS lymphoma 0.02%, GCT 0.15% as described below) (Eur J Endocrinol 2007;156(2):203)
Evaluation
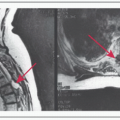
Sellar MRI to better characterize the lesion
Evaluation of hypothalamic-pituitary hormonal function
Classification: By size & cell of origin
Size: <1 cm = microadenoma; >1 cm = macroadenoma
Cell type: Arise from any type of cell of the anterior pituitary; can lead to ↑ secretion of hormone(s) produced by that cell and/or ↓ secretion of other hormones due to compression of other cell types
Gonadotroph: Usu clinically nonfunctional
Corticotroph: Usu causes Cushing
Lactotroph: ↑ PRL → hypogonadism (♂/♀)
Thyrotroph: Can be clinically nonfunctional (secreting only α or TSH-β subunits, or can cause hyperthyroidism from ↑ secretion of intact TSH)
Somatotroph: ↑ GH → acromegaly
Lactotroph/somatotroph combinations also occur, leading to sx of both
Evaluation
MRI: Best imaging procedure to evaluate sellar masses
Evaluation of hypothalamic-pituitary hormonal function:
Hormonal hypersecretion is caused only by pituitary adenomas & defines the sellar mass as such
Tx
Gonadotroph/other clinically nonfunctioning adenomas: If large enough to cause neuro sx (visual field abnormalities, etc.), TSS is 1st line; if no neuro sx, can consider TSS if extrasellar extension present (ie, elevating optic chiasm) or monitor w/q6-12 mos exams/MRIs. Post-op monitoring is w/MRI; if residual tissue grows progressively, consider repeat surgery or XRT.
Corticotroph: Complete removal of tumor via transsphenoidal adenectomy or anterior pituitary resection is 1st line; repeat surgery or XRT for persistent/recurrent disease; medical Rx for persistent/recurrent disease or while waiting for RT effect (adrenal enzyme inhibitors- ketoconazole, metyrapone; somatostatin analog- pasireotide; cabergoline). B/l total adrenalectomy can be considered in pts not cured by pituitary surgery, RT, &/or medical Rx
Lactotroph: Tx if existing/impending neuro sx due to size (ie, >1 cm), hypogonadism or other sx due to inc PRL; DA agonist (cabergoline, bromocriptine) is 1st line to ↓ tumor size & dec prl. Pts w/nl PRL & no adenoma on MR while on low-dose DA agonist for at least 2 y, suggest trial of stopping drug w/careful monitoring of PRL/MR. If drugs ineffective (ie, substantial tumor/macroadenoma remains or sx due to ↑ PRL persist after tx), consider TSS. In pts w/very large macroadenoma who have considerable residual tumor after TSS not amenable to further surgery, consider DA agonist tx and/or XRT.Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree