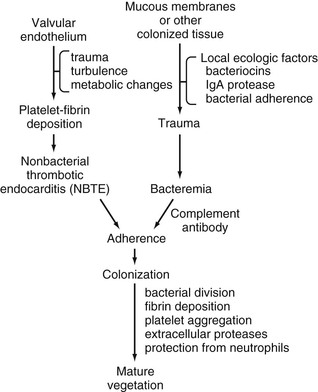
Vance G. Fowler Jr., W. Michael Scheld, Arnold S. Bayer Keywords ABE; antibiotic resistance; bacteremia; bloodstream infection; cardiac surgery; coagulase-negative staphylococci; Coxiella burnetii; culture-negative endocarditis; embolic stroke; Enterococcus; fungal endocarditis; gentamicin; HACEK; health care contact; immunopathology; infective endocarditis; injection drug use; mycotic aneurysm; nafcillin; Osler’s nodules; pathogenesis of endocarditis; petechiae; prosthetic cardiac valve; Pseudomonas aeruginosa; Roth’s spots; SBE; Staphylococcus aureus; subacute bacterial endocarditis; suppurative thrombophlebitis; vancomycin; viridans group streptococcus The term infective endocarditis (IE) denotes infection of the endocardial surface of the heart and implies the physical presence of microorganisms in the lesion. Although the heart valves are affected most commonly, the disease also may occur within septal defects or on the mural endocardium. Infections of arteriovenous shunts and of arterioarterial shunts (patent ductus arteriosus) and infections related to coarctation of the aorta are included in the following discussion because the clinical manifestations are similar. The term infective endocarditis, first used by Thayer and later popularized by Lerner and Weinstein,1 is preferable to the former term bacterial endocarditis, because chlamydiae, rickettsiae, mycoplasmas, fungi, and perhaps even viruses may be responsible for the syndrome. In the past, IE was classified as acute or subacute. This distinction was based on the usual progression of the untreated disease and is mainly of historical interest. The acute form follows a fulminant course, usually with high fever, systemic toxicity, and leukocytosis; death occurs in several days to less than 6 weeks. It classically is associated with infection caused by Staphylococcus aureus, Streptococcus pyogenes, Streptococcus pneumoniae, or Neisseria gonorrhoeae. The subacute form (death occurring in 6 weeks to 3 months) and the chronic form (death occurring later than 3 months) usually are considered together. They commonly occur in the setting of prior valvular disease and are characterized by a slow, indolent course with low-grade fever, night sweats, weight loss, and vague systemic complaints. These two forms of IE classically are caused by the viridans streptococci. Although useful conceptually, this classification ignores the nonbacterial forms of IE and the frequent overlap in manifestations of infection by specific organisms, such as the enterococci. A classification based on the etiologic agent responsible is preferable because it has implications for the course usually followed, the likelihood of preexisting heart disease, and the appropriate antimicrobial agents to employ. Although IE is relatively uncommon, it has received considerable attention from clinicians and scientists for the past century. The clinical manifestations of IE are so varied that they may be encountered in any of the medical subspecialties. Successful management depends on the close cooperation of medical and surgical disciplines. Endocarditis services and therapeutic protocols have been created at several tertiary care centers in the United States and Europe. IE has attracted considerable investigative interest. Although the factors that influence its development now are identified more clearly, many questions remain about the unique aspects of this infection, in particular: 2. What enables the microorganisms to survive on the valve surface after colonization? 3. What are the primary host defenses against induction and progression of the infection? 5. What factors are responsible for the marked variation in the manifestations of IE? These questions are discussed in detail in the following sections. The incidence of IE is difficult to determine, because the criteria for diagnosis and the methods of reporting vary with different series.2,3 An analysis based on strict case definitions often reveals that only a small proportion (approximately 20%) of clinically diagnosed cases are categorized as definite. The mean annual incidence of IE was 5 to 7 cases per 100,000 person-years in Olmsted County, Minnesota, from 1970 to 2000, with no significant change during this interval.4 A similar figure of 1.7 per 100,000 person-years was reported from a prospective survey in Louisiana,5 analogous to results from the United Kingdom6 and from France,7,8 but this figure is less than incidence reports from the Delaware River Valley region (11.6/100,000 population).9 With the use of the Nationwide Inpatient Sample (NIS), which approximates about a 20% sample of all U.S. acute care hospitals, several studies have reported significant increases in hospitalizations for IE, with most of this increase being attributable to S. aureus.10 The proportion of acute cases has increased from approximately 20% in the preantibiotic era to more than 75% in most of the industrialized world today.11 The mean age of patients with IE has increased gradually in the antibiotic era. In 1926, the median age was younger than 30 years12; by 1943, it was 39 years, and, currently, more than half of patients are older than 50 years of age.11,13,14 In a recent report, among more than 2700 patients from 58 centers in 25 countries with definite IE by the modified Duke criteria, the median age was 57.9 years.11 The disease is uncommon in children, in whom it is associated primarily with (1) underlying structural congenital heart disease, particularly septal defects or complex lesions involving septal defects; (2) surgical repair of these defects; or (3) nosocomial catheter-related bacteremia, especially in infants.15,16 The mean age for men is 6 to 7 years older than that for women, and men are affected more commonly (54% to 69% of cases); the mean male-to-female ratio is 1.7 : 1, with a range of 1 : 1 to 3 : 1 in 18 large series.17 Among patients younger than 35 years of age, more cases occur in women. Many factors may be related to this shift in age distribution. First, there has been a change in the nature of the underlying heart disease owing to a decline in the incidence of acute rheumatic fever and rheumatic heart disease countered by the increasing importance of degenerative heart disease in elderly patients. Second, the age of the population has been increasing steadily, and people with rheumatic or congenital heart disease are surviving longer. In addition, such patients increasingly are being subjected to prosthetic valve surgery, an important etiologic factor in the pathogenesis of IE. A relatively new form of the disease, health care–associated IE, has emerged secondary to the introduction of new therapeutic modalities (e.g., intravenous catheters, hyperalimentation lines, pacemakers, dialysis shunts).2,11,14,18–21 In a recent prospective, multinational cohort study of more than 1600 patients with native valve endocarditis and no injection drug use, more than one third of patients had health care–associated endocarditis, which in many cases was community acquired.21 The emerging importance of health care–associated IE in industrialized nations has also influenced the microbiology of IE, with an increasing prevalence of S. aureus and decreasing prevalence of viridans streptococci in much of the industrialized world. The heart valve involved by the infection varies considerably with the proportion of acute cases reported in each series. The distribution ranges from 28% to 45% of cases for the mitral valve alone, 5% to 36% for the aortic valve alone, and 0% to 35% for the aortic and mitral valves combined. The tricuspid valve rarely is involved (0% to 6% of cases), and the pulmonary valve even less often (<1%).11,22 Involvement of the aortic valve alone is increasing in frequency and correlates with the increase in acute cases; the incidence was 5% in 1938 and increased to 38% by 2000.11 Almost any type of structural heart disease may predispose to IE, especially if the defect results in turbulence of blood flow. Rheumatic heart disease was the underlying lesion in 37% to 76% of infections in the past, and the mitral valve is involved in more than 85% of cases related to rheumatic heart disease.17 If the mitral valve alone is involved, women outnumber men by 2 to 1. The aortic valve is affected in approximately 50% of these cases; if it alone is involved, men outnumber women by 4 to 1. Right-sided endocarditis is rare (except in injection drug users and patients with indwelling transvenous pacemakers) and accounts for fewer than 10% of all cases occurring in patients with rheumatic heart disease. In developed countries, the proportion of cases related to rheumatic heart disease has continued to decline to 5% or less in the past 2 decades,11 whereas in developing countries, rheumatic heart disease remains the most common predisposing cardiac condition.23,24 Congenital heart disease (especially patent ductus arteriosus, ventricular septal defect, coarctation of the aorta, bicuspid aortic valve, tetralogy of Fallot, and, rarely, pulmonic stenosis) is responsible in 6% to 24% of endocarditis cases.11 IE is uncommon in the secundum atrial septal defects, probably because this lesion results in a low-pressure shunt with little turbulence. The congenitally bicuspid aortic valve is an important condition in elderly patients (especially men).25 In one prospective multicenter analysis, it was present in 16% of 310 cases of definite native valve endocarditis . Half of the patients with bicuspid aortic valve had perivalvular abscess, and 72% required valve surgery.26 Surgical closure of a ventricular septal defect lowers the risk for IE.27 The degenerative cardiac lesions (e.g., calcified mitral annulus, calcific nodular lesions secondary to arteriosclerotic cardiovascular disease, post–myocardial infarction thrombus) assume greatest importance in the 30% to 40% of IE patients without underlying valvular disease. The actual contribution made by these lesions is unknown, but they occur with an increased incidence in the elderly. In one series, degenerative lesions were present in 50% of patients older than 60 years of age with native valve IE.28 The contribution of these degenerative cardiac lesions to the development of IE was apparent in an analysis of 148 patients receiving treatment in London after 1970.29,30 The underlying structural cardiac defects were as follows: rheumatic heart disease in 39 patients, congenital defects in 13, and normal or degenerate valves in 65. Although a calcified mitral annulus is fairly common in elderly women, this lesion rarely is complicated by IE (only 3 of 80 patients in one report).31 When patients with acute IE are considered separately, more than 50% have no recognized underlying cardiac disease.32 Many other conditions, such as bicuspid aortic valve,25 luetic heart disease, arterioarterial fistulas, hemodialysis shunts or fistulas, intracardiac pacemaker wires, and intracardiac prostheses, may predispose to IE. Prosthetic valve endocarditis is increasing in incidence in proportion to other forms of endocarditis. For example, it was present in one fifth of the 2781 adults with definite IE recently reported by the International Collaboration on Endocarditis-Prospective Cohort Study (ICE-PCS) (see Chapter 83).11 Intravascular infections involving cardiac devices (e.g., permanent cardiac pacemakers, defibrillators) also have increased significantly since the 1990s and are discussed in Chapter 84.33 IE also occurs more frequently among patients with extensive contact with the health care system.14,21 As noted previously, injection drug users constitute another group with an increased risk for IE (see later discussion). In this population, there is the added problem of a rapidly rising prevalence of IE among persons with human immunodeficiency virus (HIV) infection. In addition, injection drug users are the group at greatest risk for recurrent and polymicrobial IE.17,34 Although the contribution of invasive procedures (e.g., sigmoidoscopy, colonoscopy) has been debated, native valve IE seems to be more common among patients with active inflammatory bowel disease (6 of 213 patients in one report35). Although it is not classically recognized as a condition leading to bacterial endocarditis, up to 5% of patients with idiopathic hypertrophic subaortic stenosis develop IE.36 IE is more common in the subset of these patients who have hemodynamically severe forms of the disease, as manifested by a higher peak systolic pressure gradient and a high prevalence of symptoms. New murmurs develop in 36% of patients with idiopathic hypertrophic subaortic stenosis complicated by IE, and this new physical finding correlates with a higher mortality rate.36 Among seven cases examined histologically, the infection was found on the aortic valve in three cases, on the mitral valve in two cases, on both valves in one case, and on the subaortic endocardium in one case. This distribution probably is related to the associated mitral regurgitation caused by displacement of the anterior leaflet by the abnormal ventricular architecture and by the turbulence of the jet stream affecting the aortic valve distal to the intraventricular obstruction. An association has also been recognized between IE and mitral prolapse syndrome. Of 87 consecutive cases of IE reported from Stanford University, 10 (11%) occurred in patients with well-documented mitral valve prolapse.37 These 10 cases represented more than one third of the 28 cases in which isolated mitral regurgitation was the predisposing condition. Four additional cases occurred in patients who were not studied by echocardiography or angiography but who had clinical evidence of mitral prolapse syndrome. Of the cases of IE associated with isolated insufficient mitral valves, 40% to 50% probably occurred in patients with mitral prolapse syndrome. In one series of 63 cases of native valve endocarditis diagnosed in Memphis from 1980 to 1984, mitral valve prolapse was the most common underlying lesion (29%). In another study,38 5 of 58 patients with mitral valve prolapse who were monitored prospectively for 9 to 22 years developed IE. This syndrome should be suspected in patients who have midsystolic clicks with or without a late systolic murmur. The condition is common and has been recognized in 0.5% to 20% of otherwise healthy people, especially young women. It has become apparent that mitral valve prolapse is only one component of a developmental syndrome. This lesion often is associated with a distinct habitus in women,39 with von Willebrand disease, or with ophthalmoplegia. Some of these characteristics may be useful in identifying patients at high risk for IE. All 25 patients who developed IE on a prolapsing mitral valve had a holosystolic murmur, and none had the isolated click without a murmur.37 The risk for IE seems to be increased in the subset of patients with mitral valve prolapse who exhibit thickened leaflets with valvular redundancy.28 In addition, men older than 45 years of age who have mitral valve prolapse are at increased risk for IE.40 Nevertheless, the risk for IE is higher in patients with mitral valve prolapse. In a careful retrospective, epidemiologic, matched case-control analysis, the calculated odds ratio (8.2; 95% confidence interval, 2.4 to 28.4) indicated a substantially higher risk for the development of IE in these patients than in controls.41 It seems that, when IE develops in people with mitral valve prolapse, the symptoms and signs are more subtle and the mortality rate is less than in left-sided IE of other types.42 In vitro observations and studies in experimental animals have shown that development of IE probably requires the simultaneous occurrence of several independent events, each of which may be influenced by a host of separate factors. The valve surface first must be altered to produce a suitable site for bacterial attachment and colonization. Valve surface changes may be produced by various local and systemic stresses, including blood turbulence and the offending organism itself. These alterations result in the deposition of platelets, fibronectin, fibrin, and other matrix ligands in the formation of so-called sterile vegetation—the lesions of nonbacterial thrombotic endocarditis (NBTE). Bacteria then must reach this site and adhere to and invade the involved tissue to produce colonization and persistence. Certain strains seem to have a selective advantage in adhering to platelets, fibronectin, or fibrin and produce the disease with a lower inoculum. After colonization, the surface is covered rapidly with a protective sheath of fibrin and platelets to produce an environment conducive to further bacterial multiplication and vegetative growth. The interaction of these events is depicted in Figure 82-1. In the following sections, these factors are considered independently (for in-depth discussions, see references 43–46). Luschka, in 1852, first suggested that IE resulted when septic coronary emboli lodged in the vessels of the cardiac valve.47 This hypothesis was discarded, because cardiac valves are poorly vascularized.45,48,49 It now is clear that the initial colonization occurs on the damaged endothelial surface of the valve. In experimental animals, it is almost impossible to produce IE with intravenous injections of bacteria, unless the valvular surface first is perturbed. If a polyethylene catheter is passed across the aortic valve of a rat or rabbit, IE is produced with intravenously injected bacteria or fungi.50,51 Microscopic examination of this early lesion shows the organisms intimately adherent to fibrin-platelet deposits overlying interstitial edema and mild cellular distortion that have formed in areas of valvular trauma.52 Scanning electron micrographs of the damaged valvular surface confirm the adhesion of microorganisms to these areas of fibrin-platelet deposition early in the disease course.53 The organisms are covered rapidly by fibrin.54 Opossums and pigs are the only animals known to develop IE readily (i.e., without experimentally induced valvular alteration).46,55 The stress of captivity is apparently sufficient in these animals to produce subtle valvular changes that lead to spontaneous endocarditis and a markedly increased susceptibility to the disease after intravenous injection of bacteria. In other animals and probably in humans, alteration of the valve surface is a prerequisite for bacterial colonization. Angrist and Oka48 first recognized the importance of these deposits as the crucial factor in allowing bacterial colonization of valve surfaces and suggested the term NBTE. Many forms of exogenous stress produce these lesions experimentally, including infection, hypersensitivity states, cold exposure, simulated high altitude, high cardiac output states, cardiac lymphatic obstruction, and hormonal manipulations.46 These procedures all increase the susceptibility of the animals to IE. NBTE has been found in patients with malignancy (particularly pancreatic, gastric, or lung carcinoma) and other chronic wasting diseases, rheumatic and congenital heart disease,48 uremia, and connective tissue diseases such as systemic lupus erythematosus (SLE) and rheumatoid arthritis; after the placement of intracardiac catheters (e.g., Swan-Ganz); and after a self-limited acute illness. NBTE generally reflects one of two pathogenic mechanisms: hypercoagulability or endothelial damage. In a careful analysis performed in Japan, NBTE was found in 2.4% of 3404 autopsies, especially in elderly people with chronic wasting disease.56 Among patients at high risk for NBTE, the rate may be greater. Cardiac valvular vegetations were found in 19% of 200 nonselected ambulatory patients with solid tumors undergoing prospective echocardiographic screening.57 Valvular lesions were most common among patients with carcinoma of the pancreas or lung or lymphoma. NBTE is most common on the low-pressure side of the cardiac valves along the line of closure, precisely the site most often involved in IE. Whether this lesion is always essential for the development of IE in humans is unknown. Secondary hypercoagulable states (e.g., as seen in secondary antiphospholipid syndromes in SLE) may contribute to the development and propagation of the NBTE lesion.58 When associated with valvular insufficiency, IE characteristically occurs on the atrial surface of the mitral valve and the ventricular surface of the aortic valve. Rodbard59 showed that this localization is related to a decrease in lateral pressure (presumably with decreased perfusion of the intima) immediately downstream from the regurgitant flow. Lesions with high degrees of turbulence (e.g., small ventricular septal defect with a jet lesion, valvular stenosis resulting from insufficient valves) readily create conditions that lead to bacterial colonization, whereas defects with a large surface area (large ventricular septal defect), low flow (ostium secundum atrial septal defect), or attenuation of turbulence (chronic congestive heart failure [CHF] with atrial fibrillation) rarely are implicated in IE. Cures of IE achieved with ligation of an arteriovenous fistula or patent ductus arteriosus without further treatment also highlight the importance of hemodynamic factors. A hyperdynamic circulation itself, such as that developing after experimentally induced arteriovenous fistulas in dogs or after creation of fistulas and shunts in hemodialysis patients, may lead indirectly to IE by producing NBTE.45,46 Finally, implantable intracardiac prosthetic material may well contribute to a turbulent blood flow state, as seen in intraventricular pacemakers and implantable cardioverter-defibrillators. The degree of mechanical stress exerted on the valve also affects the location of the IE.60 In 1024 autopsy cases of IE reviewed through 1952, the incidence of valvular lesions was as follows: mitral, 86%; aortic, 55%; tricuspid, 19.6%; and pulmonic, 1.1%. This incidence correlates with the pressure resting on the closed valve: 116, 72, 24, and 5 mm Hg, respectively. In the setting of preexistent NBTE, transient bacteremia may result in colonization of these lesions and may lead to the development of IE.61 Transient bacteremia occurs whenever a mucosal surface heavily colonized with bacteria is traumatized, such as occurs with dental extractions and other dental procedures or with gastrointestinal, urologic, or gynecologic procedures (Table 82-1).61,62 The degree of bacteremia is proportional to the trauma produced by the procedure and to the number of organisms inhabiting the surface; the organisms isolated reflect the resident microbial flora. The bacteremia usually is low grade (≤10 colony-forming units [CFU]/mL) and transient; the bloodstream usually is sterile in less than 15 to 30 minutes. TABLE 82-1 Incidence of Bacteremia after Various Procedures Data from Everett ED, Hirschmann JV. Transient bacteremia and endocarditis prophylaxis: a review. Medicine (Baltimore). 1977;56:61. In two studies in which samples for blood cultures were drawn from patients with severe gingival disease before the dental procedure, spontaneous bacteremia was identified in 9% to 11% of the patients. Other studies have shown an even higher frequency of spontaneous bacteremia. Of the blood cultured from healthy people, 60% to 80% of specimens were positive when filters and anaerobic techniques were used.63 The degree of bacteremia was low, however, with only 2 to 10 CFU/5 mL of blood isolated. “Nonpathogenic” organisms, such as Propionibacterium acnes, Actinomyces viscosus, Staphylococcus epidermidis, and other Actinomyces or streptococcal species, were responsible. Frequent episodes of silent bacteremia also are suggested by the identification of circulating humoral antibodies to the resident oral flora and by the noted increase in sensitized peripheral T cells to the flora of dental plaque. The frequency of such silent bacteremias in the probable pathogenesis of IE has contributed to the current concept that individual dental procedures are uncommonly the cause of such infections.64 Another crucial factor during the transient bacteremia stage is susceptibility of the potential pathogen to complement-mediated bactericidal activity. Only “serum-resistant,” gram-negative aerobic bacilli (e.g., Escherichia coli, Pseudomonas aeruginosa, Serratia marcescens) reliably produce experimental IE in rabbits,65,66 and this property is found in all isolates from human cases of IE. Although experimental IE can be induced in rats with “serum-sensitive” E. coli, the organisms are eliminated rapidly on catheter removal.66 The ability of certain organisms to adhere to NBTE lesions is a crucial early step in the development of IE. Gould and associates67 showed that organisms commonly associated with IE (enterococci, viridans streptococci, S. aureus, S. epidermidis, P. aeruginosa) adhered more avidly to normal canine aortic leaflets in vitro than did organisms uncommon in IE (Klebsiella pneumoniae, E. coli). In addition, S. aureus and the viridans streptococci produce IE more readily than does E. coli in the rabbit model of IE.68 This observation correlates with the relative frequency with which these organisms produce the disease in humans. The rarity of IE due to gram-negative aerobic bacilli also may be related to their serum sensitivity, as noted previously. Differences in the propensity to cause IE are apparent even within a single species. For example, specific clones of S. aureus, including clonal complexes 3069,70 and 2271 have been reported to be associated with an increased risk for IE. In addition, only 2 of the 11 capsular serotypes of S. aureus described to date, serotypes 5 and 8, account for approximately 75% of clinical isolates, whereas highly mucoid strains (e.g., serotypes 1 and 2) are rarely recovered. Nevertheless, mutant strains devoid of microencapsulation had significantly lower median infective dose (ID50) values in a rat (catheter-induced NBTE) IE model,72 compared with wild-type parent strains. Microcapsule expression may attenuate S. aureus IE production by blocking bacterial cell surface adhesins, but this hypothesis requires confirmation. In addition, an increasing number of reports suggest that other specific pathogen characteristics in S. aureus are associated with the severity of infection caused by these strains in humans.73–77 Other studies using an elegant experimental model of IE after dental extraction in rats with periodontitis, which closely resembles the presumed pathogenetic sequence in humans, also suggest an important role for bacterial adhesion to NBTE in the early events. Although viridans streptococci were isolated much more commonly than group G streptococci in blood cultures obtained 1 minute after extraction, the latter strains caused 83% of the IE episodes in this rat model.78,79 This propensity to cause IE was associated with an increased adhesion of group G streptococci to fibrin-platelet matrices in vitro.79 The adherence of oral streptococci to NBTE may depend on the production of a complex extracellular polysaccharide, dextran. This polymer plays an essential role in the pathogenesis of dental caries by Streptococcus mutans.80 Dextran allows the organism to adhere tightly to the surface of dental enamel. The enhanced ability to adhere to inert surfaces also may be important in IE. In an analysis of 719 cases of streptococcal infections in the United Kingdom, 317 cases of IE were found.81 The most common etiologic agents were Streptococcus sanguinis (16.4% of the cases), previously identified in Streptococcus subacute bacterial endocarditis, and S. mutans (14.2%). Ratios of endocarditis to nonendocarditis bacteremia caused by particular organisms have been derived (Table 82-2), allowing prediction of the relative propensity for a particular organism to cause IE. The ratios range from 14.2 : 1 for S. mutans to a reversed ratio of 1 : 32 for S. pyogenes. Only the first four organisms listed in Table 82-2 (all with ratios >3 : 1) produce extracellular dextran. This finding suggests that dextran production may be another virulence factor in the pathogenesis of IE. TABLE 82-2 Ratio of Infective Endocarditis Cases to Nonendocarditis Bacteremia Cases for Various Streptococci and Enterococci Modified from Parker MT, Ball LC. Streptococci and aerococci associated with systemic infection in man. J Med Microbiol. 1976;9:275. The role of dextran in the adherence of oral streptococci to NBTE also has been studied in vitro with the use of artificial fibrin-platelet matrices (simulating NBTE). The amount of dextran produced by organisms grown in broth correlated with adherence; the amount was increased by incubating the organism in sucrose (which stimulates dextran production) and decreased by the addition of dextranase (which removes the dextran from the cell surface). The addition of exogenous dextran to S. sanguinis grown in sucrose-free media increased adherence. Dextran production also correlated directly with the ability of these organisms to produce IE in vivo in the rabbit model.82 The strain of S. sanguinis produced IE less readily when incubated in dextranase than did control strains, and a strain that produced large quantities of dextran produced endocarditis more easily than did a strain that produced relatively small quantities of dextran. Dextran production also increased the adherence of S. mutans to traumatized canine aortic valves in vitro,83 an effect that was dependent on polymers of higher molecular weight.84 Dextran formation (or, more properly, exopolysaccharide or glycocalyx production) by oral streptococci may be a virulence factor for the production of IE by these organisms.85 Continued in vivo synthesis of exopolysaccharide during experimental IE correlated with vegetation size and resistance to antimicrobial therapy.86,87 Measurement of cell-adherent glycocalyx by a quantitative spectrophotometric tryptophan assay among viridans streptococci isolated from blood cultures has potential value as an independent predictor of the likelihood of IE.88 Non–dextran-producing streptococci may produce IE in humans and adhere to artificial fibrin-platelet surfaces in vitro,89 suggesting that other microbial surface characteristics are instrumental for this early event. Whatever the role of the extracellular glycocalyx in microbial adhesion, its presence may retard antimicrobial therapy for streptococcal endocarditis (see later discussion).86,87,90 FimA, a surface adhesin expressed by viridans streptococci, has been shown to mediate the attachment of such organisms to platelet-fibrin matrices in vitro and to experimental NBTE lesions in the animal model of IE.91 Homologues of the fimA gene are widely distributed among clinical strains of viridans streptococci and enterococci, suggesting its importance in IE.92 Several lines of experimental evidence have confirmed further the key role of FimA in the pathogenesis of IE. Inactivation of the fimA gene has yielded viridans streptococcal mutants exhibiting a significant decrease in virulence in experimental IE compared with the parental strain having intact FimA expression.91 In addition, animals either passively immunized with anti-FimA antibody or actively immunized with a FimA vaccine were significantly less susceptible to experimental IE than nonimmunized controls.92 Recent data have emphasized the unique interaction of the viridans group streptococci with human platelets in the pathogenesis of IE. The Sullam laboratory published a series of elegant experimental studies93–95 that supported the following paradigms in viridans group streptococcal IE. First, they identified a group of genes in one viridans streptococcal species (Streptococcus gordonii, formerly known as S. sanguis I) that mediates the binding of this organism to human platelets. This locus encodes GspB, a large, serine-rich cell wall–anchored glycoprotein; four proteins mediating the glycosylation of GspB; and seven proteins (the accessory Sec system) that mediate the export of GspB to the bacterial surface. GspB is the adhesin that binds platelet glycoprotein Ib, by means of its high-affinity interaction with sialic acid–containing motifs on glycoprotein Ib. GspB is an unusual protein, not only because of its size (286 kDa) but also because glycoproteins are rare in bacteria. Glycosylation is required for the stability of GspB; loss of carbohydrate results in rapid degradation. This modification, however, renders it an unsuitable substrate for export by the canonical general secretory (Sec) pathway. In contrast, the accessory Sec system is a specialized pathway whose sole function is translocation of GspB from the cytoplasm to the bacterial surface, where it is covalently linked to the cell wall by its LPRTG C terminus. Loss of GspB expression was linked with decreased virulence in experimental IE. Similar results have been reported for Hsa, a homologue in another viridans streptococcal species (S. gordonii). Second, by transposon mutagenesis, the Sullam group discovered that binding of viridans streptococci to mammalian platelets is mediated in part by two phage-encoded proteins, PblA and PblB, that are associated with the cell wall of this organism. These proteins appear to be important for virulence, in that loss of their expression was associated with decreased virulence in an animal model of IE. This was a surprising finding, in that these are two phage proteins of a lysogenic phage (SM1) that resides on the chromosome of the strain (SF100). PblA is a tape measure protein (important for phage morphogenesis), whereas PblB is a tail fiber protein. Both proteins are linked to the bacterial surface through their interactions with choline groups within the cell wall. A puzzling issue was how these phage proteins get out of the bacterium and onto its surface. It appears that there is some constitutive, low-level expression of the phage lytic cycle, which results in the expression of phage holin and lysin. The net effect is to render the bacteria more permeable, so that PblA and PblB leak out of the bacteria and then bind back to the surface, where they can serve as adhesins. This is a remarkable system, because it is perhaps the first example of a phage-encoded bacterial surface structure and adhesin and because it suggests a co-adaptation or coevolution of the bacterium and the phage, wherein the bacterium is essential for phage replication but the phage also gives the bacterium a selective advantage by providing it with an adhesin. A similar important role of adhesion to NBTE in the pathogenesis of IE has been shown for yeasts. Candida albicans adheres to NBTE in vitro and produces IE in rabbits more readily than does Candida krusei, a nonadherent yeast rarely implicated in IE in humans.96 Although microbial adhesion is a crucial early event in the pathogenesis of IE, the precise intracardiac loci are unknown and may differ among organisms. Most organisms probably adhere initially to a constituent of NBTE; some evidence implicates fibronectin as the host receptor within NBTE.97 More recent studies98,99 have supported this concept. Low-fibronectin–binding mutants of S. aureus and S. sanguinis had decreased ability to produce IE in rats, compared with high-fibronectin-binding parent strains. Other normal constituents of damaged endothelium or NBTE (e.g., fibrinogen, laminin, type IV collagen100) also may serve to bind circulating bacteria. Streptococcus defectivus, the major species isolated in cases of IE caused by nutritionally variant streptococci (discussed later), bound the extracellular matrix of fibroblasts and endothelial cells in a saturable-specific manner, whereas Streptococcus adjacens and serotype III nutritionally variant streptococcal strains did not bind.101 A study also documented binding of S. mutans, Streptococcus mitis, S. sanguinis, and Enterococcus faecalis to this extracellular matrix. Laminin-binding proteins (e.g., a 145-kDa protein found in S. gordonii) were identified on the cell walls of organisms recovered from patients with IE,102 and the level of protein expression seemed to be regulated by the presence of extracellular matrix proteins. Other organisms may bind directly to, or become ingested by, endothelial cells as the initial event.103–106 This sequence appears to be important in the initiation of IE by S. aureus on “normal” cardiac valves or on native endothelial surfaces adjacent to damaged endothelial sites. Many studies in experimental IE, using S. aureus as the study organism, have shed additional light on the importance of microbial binding to specific matrix proteins found within the NBTE lesion on the development of IE. It seems that the key adhesin possessed by the organism for induction of IE is one or more of its several fibrinogen-binding proteins (e.g., clumping factor, coagulase107,108). Adhesins for other matrix molecules (e.g., fibronectin, collagen, thrombospondin109–111) are not involved pivotally in initial attachment of the organism to damaged endothelium but are crucial in persistence of the microbe at this site. Additional virulence factors produced by this organism (α-toxin112) have been identified in the experimental IE model as important for persistence and proliferation of the organism within maturing vegetations in the next stage of infection, after valvular colonization. The fibronectin-binding adhesins of S. aureus seem to be crucial in the ability of the organism to invade cardiac endothelium and induce endothelial apoptosis,113–115 although the specific microbial surface–host receptor ligand relationship remains incompletely defined for all the major IE pathogens. This is an active area of investigation, because inhibition of these events may provide novel prophylactic strategies. The importance of adherence characteristics in the development of endocarditis also has been examined through the use of preincubation of organisms with antibiotics. Many classes of drugs, after incubation even at subinhibitory concentrations, decrease the adhesion of streptococcal species to fibrin-platelet matrices and damaged canine valves in vitro.116 Several elegant studies in animal models verified the significance of this in vitro observation: preincubation of the organism in subinhibitory antibiotic concentrations prevented the development of IE in vivo.117,118 This finding has direct relevance to the chemoprophylactic prevention of IE (see Chapter 85). In one study, subinhibitory concentrations of penicillin were found to result in a loss of streptococcal lipoteichoic acid, with reduced adhesion to NBTE-involved tissue and an impaired ability to produce IE in vivo.119 Antibiotics may prevent IE by at least two mechanisms—bacterial killing and inhibition of adhesion to NBTE-involved tissue.120 Because platelets and fibrin are the major constituents of NBTE, the role of the platelet in the pathogenesis of IE also has been studied. Some strains of bacteria have been found to be potent stimulators of platelet aggregation and the release reaction.121 In general, IE-producing strains of staphylococci and streptococci more actively aggregate platelets than do other bacteria that less frequently produce IE. Bacteria-platelet aggregates have been found in the peripheral blood of patients with bacteremia. The importance of these aggregates in the formation of the vegetation (or, conversely, the effect of the aggregation on the rate of removal of organisms from the circulation) is unknown. In one study, even small numbers of platelets greatly increased the adherence of oral streptococci to fibrin in vitro.81 Other studies122 showed that S. sanguinis, an important cause of IE, aggregates platelets and adheres to these blood components by means of protease-sensitive components, not dextrans. A platelet receptor for ligands on certain strains of S. sanguinis was suggested. However, this platelet aggregation by viridans streptococci requires direct platelet binding and plasma components.123 Other experiments implicated immunoglobulin G (IgG) in this specific streptococcal bacteria-platelet interaction and suggested that platelet activation is mediated through the platelet surface Fc receptor, with a molecular weight of 40,000 daltons.124 After colonization of the valve occurs and a critical mass of adherent bacteria develops, the vegetation enlarges by further platelet-fibrin deposition and continued bacterial proliferation. There is a complex interplay among factors responsible for bacteria-platelet adhesion and aggregation. The ability of S. sanguinis to induce platelet aggregation in vitro is conferred by two bacterial cell surface antigens: (1) class I antigen, which promotes adhesion of S. sanguinis to platelets (adh+), and (2) coexpression of class II antigen, which promotes platelet adhesion or platelet aggregation (agg+). At least nine adh/agg phenotypes have been identified among naturally occurring variants, reflecting a range of platelet interactivity. Intravenous inoculation of agg+ S. sanguinis strains into rabbits with catheter-induced aortic valve trauma led to larger vegetations, a more severe clinical course, more gross lesions in major organs, and greater mortality than inoculation with an agg− strain or with the agg+ strains pretreated with Fab fragments specific for the platelet interactivity phenotype.125 Platelet aggregation induced by S. sanguinis in vivo seems to be an important virulence determinant of vegetation development and disease progression. Streptococcal exopolysaccharide production inversely correlates with platelet adhesion while inhibiting aggregation,126 indicating that these surface molecules may enhance endocarditis at some pathogenic steps but not at others. The manner in which S. aureus interacts with platelets in the pathogenesis of IE differs substantially from that of the viridans streptococci. This interaction does not require the presence of specific antistaphylococcal antibody and is not amplified by the platelet Fc receptor.127 Platelet–S. aureus interactions for executing aggregation require fibrinogen as a bridging molecule but are independent of the primary platelet fibrinogen-binding site, the glycoprotein IIb/IIIa integrin receptor. In addition, it seems that S. aureus can bind to platelets via platelet-derived von Willebrand factor or directly to von Willebrand factor receptor, at von Willebrand factor–binding domain.128–130 In addition, platelet surface–expressed gCiqR can serve as a key “docking site” for staphylococci, predominantly through bridging molecules such as protein A and von Willebrand factor.131 In experimental IE, transposon inactivation of the putative S. aureus platelet-binding adhesin gene resulted in mutants with diminished capacity to adhere to platelets in vitro in either suspension or surface-bound monolayers.132 In experimental IE caused by such low-platelet–binding mutants, the induction rates of IE were equivalent to the induction rates of the parental strain, presumably because of the microbe’s ability to attach to damaged endothelium by multiple adhesive mechanisms. However, the capacity of the mutant to persist and proliferate within experimental vegetations and to disseminate hematogenously to the kidneys was markedly impaired in the mutant strain.132 This transposon defect was found to reside within the staphylococcal fibrinogen adhesin gene, clumping factor A (clfA).133 Platelets also may play a role in host defense within the cardiac vegetation during IE.134 It is underappreciated that platelets can actually phagocytose circulating staphylococci into engulfment vacuoles, in which the organism can persist.135 Moreover, after specific exposure to thrombin (which is plentiful at the surface of damaged endothelium), release of α-granule–derived platelet microbicidal proteins (PMPs), or thrombocidins, with bactericidal activity against most gram-positive cocci that cause IE, has been shown.136 PMPs appear to be homologues of platelet factor 4,137 whereas thrombocidins evolve from the platelet basic peptide lineage and are truncates of the chemokines NAP-2 and CTAP-3.138 Although the ability of S. aureus to adhere to and aggregate platelets is a related property, the resistance to PMPs is an independent phenotypic characteristic and a potential virulence factor.139 PMPs are low-molecular-weight (8 to 10 kDa) cationic proteins and may act primarily on the bacterial cell membrane or cell wall, synergistically with antibiotics, to kill bacteria. PMPs also have shown fungicidal activity against some yeasts in vitro.140 Microbial resistance to the cidal activity of PMPs may contribute to the pathogenesis of IE. This hypothesis was supported by a reduction in vegetation weight and bacterial concentration in rabbits with experimental aortic valve S. aureus endocarditis after treatment with aspirin.141 In addition, three studies in experimental IE confirmed the importance of the relationship of PMP resistance and the pathogenesis of IE. In experimental viridans streptococcal IE and S. aureus IE, PMP-resistant strains exhibited an enhanced capacity to persist at sites of valvular damage.142,143 In addition, S. aureus strains exhibiting the PMP resistance phenotype in vitro were able to proliferate within the vegetation and hematogenously seeded extracardiac foci (kidneys, spleen) to a significantly greater extent than their isogenic counterparts, which were PMP susceptible in vitro.144,145 Several clinical studies also emphasized the important association between PMP resistance and the pathogenesis of intravascular infections. Bloodstream isolates of viridans streptococci and S. aureus from patients with IE tended to be substantially more resistant to PMPs in vitro.143 S. aureus bloodstream isolates arising from an intravascular source (catheter or IE) were significantly more resistant than bloodstream isolates arising from a deep tissue source.146 Further, PMP-resistant S. aureus bloodstream isolates from patients with IE were significantly more likely to have arisen from an intravascular catheter source.147 Lastly, recent data have shown fusion of PMP-rich α-granules with platelet engulfment vacuoles noted earlier, implicating the intrinsic PMP-resistance phenotypes of engulfed staphylococci in their ultimate intraplatelet survival outcomes.135 Should the engulfed organism be PMP resistant, the platelet could serve as a “Trojan horse” vehicle to disseminate the organism. In contrast, if the phagocytosed staphylococci were PMP susceptible, this mechanism could eliminate the organism from the circulation.135 In IE, the bacterial colonies are found beneath the surface of the vegetation (at variable depth, depending on the intracardiac location148) and infiltration by phagocytic cells is minimal; the vegetation creates an environment of impaired host resistance. These conditions allow for relatively unbridled bacterial growth, resulting in extremely high colony counts of 109 to 1011 bacteria per 1 g of tissue. Bacteria deep within the fibrin matrix have been shown by autoradiography to reach a state of reduced metabolic activity.149 Studies by Yersin150 and Meddens151 and their colleagues suggested that impairment of host defenses (e.g., neutropenia, corticosteroids) potentiates progression of the disease when the tricuspid, but not the aortic, valve is involved but is largely dependent on the intracardiac location of the vegetation.152 The role of granulocytes within the vegetation is unknown. When vegetation formation is retarded with anticoagulants in experimental animals with IE, the organisms seem to divide on the surface, total bacterial titers are lower, and the clinical disease is more explosive.153,154 In addition, it has been suggested that phagocytosis of microorganisms by monocytes on or within the vegetation generates tissue thromboplastin formation; thromboplastin then acts as a stimulant to fibrin deposition and growth of the vegetation.155 However, the best evidence suggests that coagulation activation initiated by tissue factor,156 with subsequent local thrombus formation, is responsible for the initiation of vegetation growth and persistence on the cardiac valve. It seems that some organisms (e.g., S. aureus) induce tissue factor production by endothelium without the necessity for host cytokines.157 Many important studies have elucidated the interactions among the invading microbe, the endothelium, and the monocyte in the pathogenesis of IE. After internalization by endothelial cells in vitro, microbes such as S. aureus evoke a potent proinflammatory chemokine response, including, for example, increased expression of interleukin-6 (IL-6) or IL-8 or of monocyte chemotactic peptide.158,159 This event also is associated with increased expression on the endothelial cell surface of several key adhesion molecules, especially intercellular adhesion molecule 1 (ICAM-1) and vascular cell adhesion molecule 1 (VCAM-1).106,158,159 Among other cells, monocytes are drawn into this endothelial cell microenvironment; via their appropriate counter-receptors, monocytes can bind avidly to such microbe-activated endothelial cells.159 Extracellular bacteria circulating in the vascular system then bind directly to the monocyte surface, inducing the release of tissue thromboplastin (tissue factor).160,161 This latter molecule participates in the catalytic conversion of prothrombin to thrombin, amplifying the procoagulant cascade at the site of endothelial cell colonization and leading to progressive evolution of the vegetative lesion in IE. Several studies have emphasized that organisms with low protease production (e.g., enterococci) are associated with larger, more friable vegetations with an increased tendency for embolization. This property was underscored in a recent elegant animal study by inducing IE with an enterococcus with low proteolytic activity and virulence to analyze the role of host proteases in vegetation growth. Matrix metalloprotease 9, elastase, and plasminogen activators were all present at higher concentrations in septic vegetations. These results suggest that the continuous attractant signals coming from bacterial colonies can result in chronic injury of myocardial tissues by host proteases. IE results in stimulation of humoral and cellular immunity, as manifested by hypergammaglobulinemia, splenomegaly, and the presence of macrophages in the peripheral blood. The possibility that preformed antibody can increase the likelihood of the development of IE was suggested by the spontaneous occurrence of IE in horses receiving repeated immunizations with live pneumococci.162 It was suggested that these antibodies produced bacterial agglutination in vivo, which increased the chances of valvular colonization. Studies in animals have suggested a protective role for circulating antibody. Rabbits preimmunized with heat-killed streptococci plus Freund’s adjuvant had a significantly higher ID50 than that noted for nonimmunized controls after aortic valve trauma.163 Other studies yielded similar results with S. sanguinis, S. mutans, and S. pneumoniae.164,165 In additional experiments, antibody directed against cell-surface components (including mannan) reduced the adhesion of C. albicans to fibrin and platelets in vitro and reduced IE production in vivo.166 This effect may depend on the infecting organism, however, because antibody to S. epidermidis or S. aureus did not prevent the development of IE in immunized animals and did not result in reduced bacterial concentrations in infected vegetations or kidneys,167 perhaps because of the inability of immune sera to enhance opsonophagocytosis of staphylococci. The role of preformed antibody in the pathogenesis of IE is unclear. Intravascular agglutination of bacteria may decrease the frequency of IE by reducing the actual number of circulating organisms, but cross-protection was not achieved by passive transfer of high-titer immune globulin from S. defectivus–immunized rabbits to control animals.165 Nitrogen mustard–treated immunized rabbits lost their ability to clear S. defectivus efficiently from the circulation, a process partially restored by neutrophil transfusion.168 The role of the glycocalyx of S. aureus, and of antibodies directed against this exopolysaccharide, in the pathogenesis of IE is controversial. Most experimental studies suggest that microencapsulation of strains by the common capsular types (5 and 8) may mitigate virulence of the organism in IE by obscuring key surface-expressed adhesins involved in colonization or persistence at endovascular damage sites.169 Several more recent studies suggested a salutary effect of active or passive immunization strategies against this glycocalyx in diminishing the induction, progression, or metastatic infection phases of experimental IE.170,171 However, large clinical trials have not been able to document salutary outcomes using active or passive immunization strategies directed against the staphylococcal capsule or candidate surface adhesins (e.g., clumping factor).172–175 Rheumatoid factor (anti-IgG IgM antibody) develops in about 50% of patients with IE of longer than 6 weeks’ duration.176 Rheumatoid factors were found at the time of admission in 24% of patients with acute staphylococcal IE (<6 weeks’ duration), and the frequency increased to 40% if fever persisted for 2 weeks after the initiation of antibiotic therapy.177 More than two thirds of the patients became seronegative after 6 weeks of therapy, and two patients with a second episode of acute IE promptly redeveloped positive rheumatoid factors. The titers correlated with the level of hypergammaglobulinemia and decreased with therapy. Rheumatoid factor may play a role in the disease process by blocking IgG opsonic activity (i.e., by reacting with the Fc fragment), stimulating phagocytosis, or accelerating microvascular damage. Rheumatoid factor (IgM) has not been eluted from the immune complex glomerulonephritis associated with IE.178 Antinuclear antibodies also occur in IE and may contribute to the musculoskeletal manifestations, low-grade fever, or pleuritic pain.179 Similar to malaria, schistosomiasis, syphilis, kala-azar, and leprosy, IE is associated with a constant intravascular antigenic challenge, and the development of several classes of circulating antibody is not unexpected. Opsonic (IgG), agglutinating (IgG, IgM), and complement-fixing (IgG, IgM) antibodies and cryoglobulins (IgG, IgM, IgA, C3, fibrinogen); various antibodies to bacterial heat-shock proteins; and macroglobulins all have been described in IE.180–182 Circulating immune complexes have been found in high titers in almost all patients with IE.183 Circulating immune complexes are found with increased frequency in connection with a long duration of illness, extravalvular manifestations, hypocomplementemia, and right-sided IE. Levels decrease and become undetectable with successful therapy. Patients with IE and circulating immune complexes may develop a diffuse glomerulonephritis that is analogous to the nephritis seen with infected ventriculoatrial shunts.184 Corticosteroids have been used in a few patients with glomerulonephritis associated with IE.185 Immune complexes plus complement are deposited subepithelially along the glomerular basement membrane to form a “lumpy-bumpy” pattern. Immune globulin eluted from these lesions has been shown to cross react with bacterial antigens.186 In addition, bacterial antigens have been shown within circulating immune complexes.187 Some of the peripheral manifestations of IE, such as Osler’s nodes, also may result from a deposition of circulating immune complexes. Pathologically, these lesions resemble an acute Arthus reaction. The finding of positive culture aspirates in Osler’s nodes188 suggests, however, that they may be caused by septic emboli rather than immune complex deposition. In some diffuse purpuric lesions in IE, immune complex deposits (IgG, IgM, and complement) have been shown in the dermal blood vessels by immunofluorescence.189 Quantitative determinations of serum immune complex concentrations are useful in gauging the response to therapy. Effective treatment leads to a prompt decrease, with eventual disappearance of circulating immune complexes.190 Conversely, therapeutic failures or relapses are characterized by rising titers or a reappearance of circulating immune complexes.191
Endocarditis and Intravascular Infections
Infective Endocarditis
Epidemiology
Pathogenesis and Pathophysiology
Nonbacterial Thrombotic Endocarditis
Hemodynamic Factors
Transient Bacteremia
PROCEDURE/MANIPULATION
% POSITIVE BLOOD CULTURES
Dental
Dental extraction
18-85
Periodontal surgery
32-88
Chewing candy or paraffin
17-51
Tooth brushing
0-26
Oral irrigation device
27-50
Upper Airway
Bronchoscopy (rigid scope)
15
Tonsillectomy
28-38
Nasotracheal suctioning/intubation
16
Gastrointestinal
Upper gastrointestinal endoscopy
8-12
Sigmoidoscopy/colonoscopy
0-9.5
Barium enema
11
Percutaneous needle biopsy of liver
3-13
Urologic
Urethral dilation
18-33
Urethral catheterization
8
Cystoscopy
0-17
Transurethral prostatic resection
12-46
Obstetric/Gynecologic
Normal vaginal delivery
0-11
Punch biopsy of the cervix
0
Removal/insertion of intrauterine (contraceptive) device
0
Microorganism–Nonbacterial Thrombotic Endocarditis Interaction
ORGANISM
ENDOCARDITIS : NONENDOCARDITIS RATIO
Streptococcus mutans
14.2 : 1
Streptococcus bovis I
5.9 : 1
Dextran-positive Streptococcus mitior
3.3 : 1
Streptococcus sanguinis
3 : 1
S. mitior
1.8 : 1
Unclassified viridans streptococci
1.4 : 1
Enterococcus faecalis
1 : 1.2
Miscellaneous streptococci
1 : 1.3
S. bovis II
1 : 1.7
Streptococcus anginosus
1 : 2.6
Group G streptococci
1 : 2.9
Group B streptococci
1 : 7.4
Group A streptococci
1 : 32
Immunopathologic Factors
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Endocarditis and Intravascular Infections
82