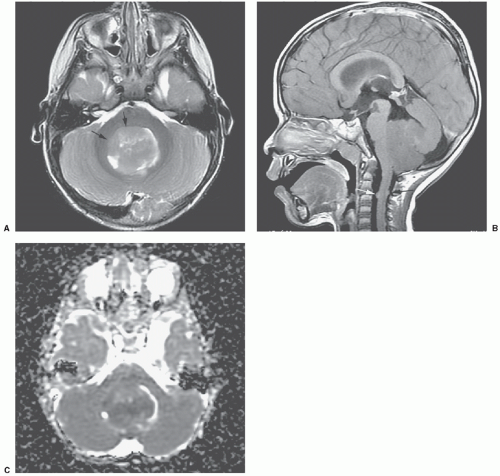
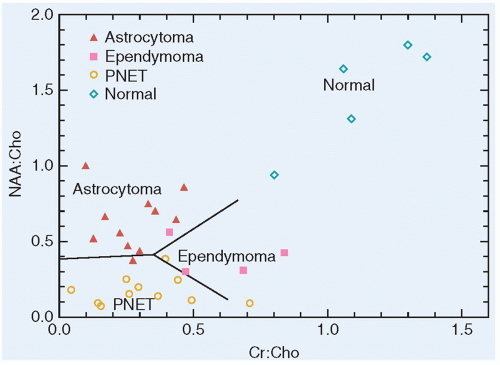
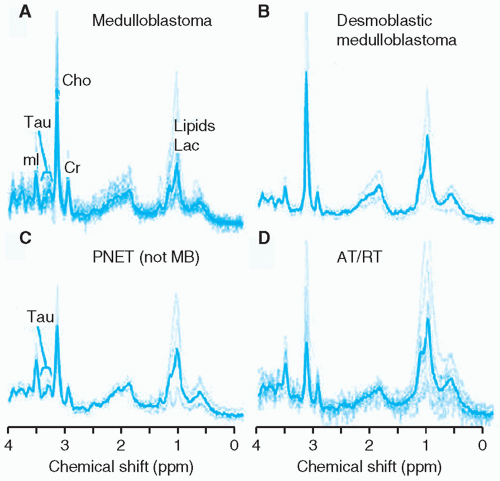
Embryonal tumors constitute approximately 25% of all primary central nervous system (CNS) tumors occurring in patients 18 years or younger,
1,2,3 and nearly 40% of all malignant CNS neoplasms. Although embryonal tumors occur throughout the pediatric age spectrum, they often arise early in life, with one-fifth or more diagnosed in the first 3 years, making diagnosis and management even more challenging.
1,2,3,4 In the World Health Organization (WHO) classification of CNS tumors, embryonal tumors constitute a collection of biologically heterogeneous lesions that are malignant and share the tendency to disseminate in the nervous system via the cerebrospinal fluid (CSF) pathways early in the course of illness.
1 The most common embryonal tumor is medulloblastoma (
Table 26B.1), with others including CNS primitive neuroectodermal tumors (PNETs) and atypical teratoid/rhabdoid tumors.
1,5,6Tumors of the pineal region are considered a distinct subset of neoplasms in the WHO classification, although they are composed of a number of biologically and clinically different tumor types, including pineoblastoma, which histologically most closely resembles embryonal tumors, such as medulloblastoma
1,7 (
Table 26B.2). Although primary CNS germ cell tumors may occur anywhere within the neuroaxis, they most commonly arise in the pineal region; and for this reason, the subgroup of germ cell tumors will also be discussed in this chapter.
1,8 General considerations and the approach to a child with a brain tumor is covered separately in Chapter 26A.
Pathologic Classification of Medulloblastoma
PNETs arising in the cerebellum are termed medulloblastoma and are classified into five histologic groups in the current WHO classification of CNS tumors
1: classic, desmoplastic nodular, medulloblastoma with extensive nodularity (MBEN), anaplastic, and large cell.
1,56 Of note, there is significant morphologic overlap
between PNETs arising in different regions of the CNS as well as in extra-CNS locations. The nomenclature used by the WHO classification relies on both the location of the tumor and the histologic appearance to render a diagnosis.
Classic medulloblastomas are highly cellular, soft, friable tumors composed of cells with deeply basophilic nuclei of variable size and shape, little discernible cytoplasm, and, often, abundant mitoses (
Fig. 26B.5) although they may exhibit surprising histologic heterogeneity. Homer Wright rosettes and pseudorosettes are variably present. Various degrees of glial or neuronal differentiation are noted, suggesting that the primitive cell of origin possesses the capacity for bipotential differentiation. A histologic variant with an abundant stromal component,
desmoplastic nodular medulloblastoma (
Fig. 26B.6), occurs dominantly in the lateral cerebellar areas of adolescents and adults and in the setting of Gorlin syndrome in infants and young children.
35,36 These may have a nodular appearance. A variant of desmoplatic nodular medulloblastoma, called
medulloblastoma with extensive nodularity (MBEN), occurs predominantly in very young children.
Aggressive variants of medulloblastoma, termed
large-cell and
anaplastic (frequently grouped together as large-cell anaplastic), have also been described. As the names imply, the histologic features that distinguish this subset of medulloblastoma are large round nuclei with prominent nucleoli, frequent mitoses, abundant apoptosis, and, in the anaplastic subset, nuclear pleomorphism. These tumors typically express synaptophysin and chromogranin. Monosomy 22 has not been seen in the cases described, but they tend to be associated with
MYC or
MCYN amplification.
57 The large-cell anaplastic variant represented 4% of the nearly 500 cases of medulloblastoma reviewed by Brown et al.
58,59
Biologic Classification and Prognosis
Correlations have been noted between histologic subtypes and clinical outcomes, prominently including an improved prognosis for nodular desmoplastic medulloblastoma and a poor prognosis for large-cell anaplastic tumors. More recently, genomic methods have enabled the identification of biologically distinct subsets of medulloblastoma and provided a biologic basis for those clinical observations. These studies have led to a current consensus that medulloblastoma consists of four molecular subtypes with characteristic genetic, demographic, and clinical features: Wnt, SHH, Group 3 and Group 4 (
Table 26B.4).
60,61,62 Groups 3 and 4 medulloblastomas are frequently categorized together as “non-Wnt, non-SHH” tumors. Retrospective analyses have revealed this classification to have important prognostic significance in patients with medulloblastoma. Patients with tumors demonstrating Wnt pathway activation have an overall survival rate greater than 90%, while SHH subtype and Group 4 tumors have an intermediate survival of approximately 75% and Group 3 tumors have the poorest outcome with overall survival ranging from 40% to 60%.
62 Classification of medulloblastomas into Wnt, SHH and non-Wnt, non-SHH subtypes (which can be performed using immunohistochemical methods on Formalin-Fixed Paraffin-Embedded [FFPE] specimens) has facilitated the subtyping of medulloblastomas for the conduct of future prospective clinical trials.
63
Wnt Subtype
Medulloblastomas with activation of the Wnt signaling pathway constitute approximately 10% of medulloblastomas overall and have the best prognosis (>90% survival) (
Fig. 26B.4). This subtype is most common in older children and adolescents (median age of 10 years) and is generally associated with tumors with classic histology. Greater than 90% of Wnt medulloblastomas have mutations in the β-catenin gene (
CTNNB1) that result in a mutant protein that is resistant to degradation and accumulates in the nucleus of tumor cells. These tumors consistently show deletion of one copy of chromosome 6, allowing monosomy 6 and nuclear β-catenin accumulation to serve as specific and sensitive markers for Wnt pathway medulloblastoma. Mutations of the
DDX3X gene are also frequent, occurring in approximately 50% of Wnt tumors.
64,65,66,67,68 Of note, mutations in
TP53, although associated with a poor prognosis in SHH pathway tumors, do not have a significant effect on the prognosis for this group of patients.
69
SHH Subtype
Approximately 30% of medulloblastomas are defined by upregulation of the SHH signaling cascade through a variety of genetic mechanisms: most frequently, mutation of the
PTCH1 tumor suppressor gene (a negative regulator of SHH signaling) and less often, alterations of other pathway members such as
SMO, SUFU, and
GLI2 (
Fig. 26B.4). Bimodal peaks of SHH medulloblastoma incidence are seen: the first in children less than 3 years of age, and the second in older adolescents and young adults. Mutations in the
SUFU gene are more common in tumors occurring in children less than 3 years of age, and
SMO mutations are more frequent in adult tumors, which have potential implications for the use of molecularly-targeted therapies targeting SHH tumors in those populations. Although the prognosis for the group of patients with SHH tumors is intermediate (˜75%),
TP53 gene mutation in an SHH pathway tumor carries a particularly poor prognosis and
may frequently be a germ line rather than somatic (e.g., in a child with Li-Fraumeni syndrome).
70 Although SHH tumors do occur in the midline location, a cerebellar hemispheric location almost always indicates a tumor of SHH subtype.
Group 3 Subtype
This subtype of medulloblastoma accounts for approximately 25% of tumors and has both the highest rate of metastases and the worst survival rate (˜50%). Unlike Wnt and SHH medulloblastomas, no defining pathway alterations have been identified, although recurrent amplifications of the MYC and OTX2 genes and mutations of SMARCA4 are seen. Group 3 medulloblastomas occur more commonly in males (male:female ratio of 2:1) and primarily affect infants and young children rather than teenagers.
Group 4 Subtype
This subtype of medulloblastoma accounts for approximately 35% of tumors and has an intermediate survival rate (˜75%) although metastatic disease is relatively frequent (˜35%). As for Group 3 tumors, the biologic basis of Group 4 tumors is poorly understood, with amplification of MYCN found in a minority of tumors and no recurrent mutations occurring in more than 10% to 15% of tumors. Isochromosome 17q is seen more commonly in Group 4 than in Group 3 tumors and is rarely seen in Wnt or SHH subtypes. Group 4 tumors are seen in children of all ages but relatively rarely in infants. Like Group 3 medulloblastoma, Group 4 tumors occur more frequently in males (male:female ratio of 3:1).
Additional genetic and biologic studies are required to further clarify the basis of medulloblastoma, and in particular Group 3 and Group 4 tumors. Future prospective studies will incorporate this subtype information for risk stratification, treatment assignment, and selection of agents to target specific subtypes of medulloblastoma with the ultimate goal of improving outcomes and decreasing treatment-related morbidities.
Staging and Risk Stratification
Although the future holds great promise with the possible incorporation of the biologic findings described earlier, clinical studies so far have relied on clinical staging and risk stratification.
4 Staging remains predominantly based on determination of the extent of tumor at the time of diagnosis and the degree of surgical resection. Complete staging requires pre- or postoperative MRI of the entire brain and spine, postoperative MRI of the tumor site, and CSF analysis.
14As with other CNS tumors of presumed primitive neuroepithelial origin, widespread seeding of the subarachnoid space may occur. The reported frequency of CNS spread outside the area of the primary tumor at diagnosis is 11% to 43%, and such spread eventually occurs in as many as 93% of patients who come to necropsy.
71,72Extraneural spread has been observed in 20% to 35% of patients in smaller institutional studies, but more recent larger series suggest that the rate of such events is less than 4%. Bone and bone marrow are the most common extraneural sites, accounting for more than 80% of extraneural metastases; lymph nodes, liver, and lung are the other reported sites.
73,74,75,76Historically, the Chang staging system, utilizing preoperative imaging studies, the surgeon’s intraoperative impression, and CSF cytology, was used to assign the stages of primary (T stage) and metastatic (M stage) disease
14 (
Table 26B.5). This system has been significantly modified and the assessment of the preoperative size of the tumor (T stage) has been supplanted by a postoperative evaluation of the extent of resection or, more specifically, the amount of postoperative residual disease, at times, modified by the impression of the surgeon at the time of diagnosis (
Table 26B.6).
The M stage of the disease remains the single, most prognostic clinical parameter.
77,78 The prognostic significance of M1 disease, indicating positive CSF cytology, without radiographic evidence of disseminated disease, has been debated and has been found to be predictive in some, but not all, studies. Part of this difference may be due to the means to assess the presence of free-floating tumor cells. Lumbar CSF cytology has been shown to be the most sensitive, but in some studies, either ventricular and/or spinal fluid have been used for assessment. Gajjar et al.
79 found lumbar CSF to be more sensitive in the detection of disseminated disease than ventricular fluid and found cytologic assessment to be complementary to neuroaxis neuroimaging. In the German HIT 1991 study, 13% of patients had M1 disease and their overall survival (OS) rate of 65% did not differ from those with M0 disease.
77 A similar finding was noted for a small number of patients with M1 disease in the French M7 study.
78 In contradistinction, the Children’s Cancer Group (CCG)-921 study found that 18% of patients had M1 disease, utilizing almost exclusively lumbar CSF, and found a 5-year and progression-free survival of 57% for those patients with positive cytology, a rate lower than that for those with M0 disease, and higher than that for patients with M2 or M3 disease, although differences were of borderline significance.
80CSF for staging is most predictive if obtained 2 weeks or more following tumor resection.
4,79 Preoperative lumbar puncture may be contraindicated if there is mass effect of the tumor and the potential for cerebellar herniation. Cisternal fluid, obtained at the time of diagnosis, has been variably related to outcome. Arachnoid biopsy, at the time of surgery, is presently being evaluated as a possible independent prognostic factor.
Neuroradiographically confirmed metastatic disease (M2 or M3) has been prognostic in all series.
77,78,79,80,81 Extraneural spread at diagnosis (M4 disease) is rare outside of infancy, and assessment for extraneural disease by bone scans and bone marrow analysis is now not recommended, in most prospective studies, as part of initial staging. The sensitivity of MRI for the detection of metastatic disease is much greater than that of CT or myelography. Because of this, it is likely that patients who in the past would have been
diagnosed as nonmetastatic, or with M1 disease, are now being classified as having M2 disease.
Despite the accepted use of MRI for staging, performance and interpretation of neuroimaging studies for the evaluation of disease dissemination at the time of diagnosis remains imperfect. In a recent Children’s Oncology Group (COG) study and French prospective studies, upon central review, inadequate imaging studies, due to patient movement or incomplete staging, were noted in up to 10% of patients.
17 Another 5% to 10% of patients, upon central neuroimaging review, were found to have significant residual postoperative disease or evidence of dissemination not appreciated by the treating institutions. Patients with evidence of dissemination, upon central review, had a particularly poor prognosis if treated on reduced-radiotherapy protocols. Patients with inadequate imaging had intermediary outcomes between those with evidence of disease dissemination and those with no evidence of disseminated disease upon central review. Determination of the extent of disease on neuroimaging studies is particularly challenging in those with nonenhancing tumors, as spinal metastasis can be quite difficult to appreciate. Similarly, spread to the third ventricle and cisterns around the brain stem was problematic to appreciate.
Extent of Resection
A near-total (arbitrarily defined as more than a 90% resection) or a total resection is now achieved in approximately 80% of children with medulloblastoma, utilizing contemporary microsurgical techniques, entered in cooperative group studies.
82 Evidence suggesting that the extent of resection correlates with outcome has been provided by several single institutions and by multi-institutional studies.
83,84 For example, in the series reported by Jenkin, a 93% 5-year progression-free survival rate was noted in children undergoing gross-total resection as comparedwith only 45% in those undergoing a partial resection.
85 In one CCG study, a clear relationship was found between the presence of greaterthan-1.5-cm
2 residual disease on postoperative imaging and a significantly lower progression-free survival rate in patients with nonmetastatic disease.
80,83 Arbitrarily, most cooperative group studies have continued to utilize some measure of the extent of residual disease after surgery as a staging criteria, as children with less than 1.5 cm
2 of postoperative residual disease have been included in the average-risk category, while those with a greater amount of disease are classified as high-risk. It is widely accepted that patients who undergo only a minimal degree of resection fare poorly.
Brain stem involvement at the time of diagnosis was found in older studies, in which patients were predominantly treated with radiation therapy alone, to be predictive of poor outcome.
9,80 However, in most studies that have coupled chemotherapy with radiation therapy, the presence of brain stem involvement has not been found to be a significant prognostic variable.
20,77,86
Histology
The significance of different histologic patterns on survival has been variable. In infants, the desmoplastic variant of medulloblastoma has been related to improved survival.
11,12 More recently, the anaplastic or large-cell variant had been linked with more extensive and/or disseminated tumors and poorer prognosis, even in cases without dissemination.
12,22,44 This latter finding has been demonstrated predominantly by retrospective reviews and in at least one prospective study.
93 In the most recent COG prospective national studies, patients with nonmetastatic disease but with extensive or “diffuse” anaplasia have been included on high-risk protocols. Most of the children with large-cell anaplastic medulloblastoma fall into the Group 3 and 4 subtypes and tend to have a poorer prognosis. The significance of anaplasia as an outcome measure has been confounded by its subjectivity and relationship to biologic markers (such as higher MYC expression) that have been related to poorer prognosis.
22