Dyslipidemias have been commonly described in patients with chronic kidney disease (CKD) since the 1960s, with case reports demonstrating markedly elevated levels of cholesterol in nephrotic syndrome patients and hypertriglyceridemia in patients with renal insufficiency. The true spectrum of dyslipidemias in kidney disease has only recently become evident, as we have gained a better understanding of changes in metabolism with mild to moderate and severe kidney disease and also the extent of kidney disease in the United States.
Currently in the United States, over 500,000 patients require dialysis or have a kidney transplant as treatment for end-stage renal disease (ESRD) (1). ESRD, however, represents only a small proportion of patients with CKD, as there are an estimated 26.3 million in the United States with mild to moderate decreases of kidney function, a number that is steadily rising with the increasing prevalence of hypertension and diabetes (2). The prevalence of intermediate stages of CKD, though not well documented, is considerably less in children and this is reflected in a U.S. prevalence of 7,500 cases of pediatric ESRD, much lower than among adults (1).
To understand the spectrum and the progression of kidney disease, a staging process has been suggested and utilized in the kidney community. This standardization of kidney function can allow for risk stratification for progression of disease. The stages are defined based on standard criteria established by the Kidney Disease Outcome Quality Initiative (KDOQI) with the stages of CKD from mild to severe disease based on estimated glomerular filtration rate (GFR) using serum creatinine-based equations. The stages are as follows: stage 1, persistent albuminuria with GFR >90 mL/min per 1.73 m2; stage 2, persistent albuminuria with GFR of 60 to 89 mL/min per 1.73 m2; stage 3, GFR of 30 to 59 mL/min per 1.73 m2; stage 4, GFR of 15 to 29 mL/min per 1.73 m2 (2). The estimated prevalence of kidney disease in the United States over time and by kidney function stages is summarized in Table 11.1.
TABLE 11.1 PREVALENCE OF CKD STAGES IN U.S. ADULTS AGED 20 YEARS OR OLDER BASED ON NHANES 1988-1994 AND NHANES 1999-2004
Prevalence ratio for NHANES 1999-2004 to 1988-1994 (95% CI)
Estimated no. of U.S. adults in 2000, no. in millions (95% CI)
NHANES 1988-1994
NHANES 1999-2004
1
1.71 (1.28-2.18)
1.78 (1.35-2.25)
1.05 (0.85-1.30)
3.6 (2.7-1.5)
2
2.70 (2.17-3.24)
3.24 (2.61-3.88)
1.21 (1.03-1.41)
6.5 (5.2-7.8)
3
5.42 (4.89-5.95)
7.69 (7.02-8.36)
1.42 (1.25-1.62)
1.55 (14.1-16.8)
4
0.21 (0.15-0.27)
0.35 (0.25-0.45)
1.70 (1.11-2.51)
0.7 (0.5-0.9)
5
NA
NA
NA
NA
Total
10.03 (9.16-10.91)
13.07 (12.04-14.10)
1.30 (1.19-1.43)
26.3 (24.2-28.3)
aDefined based on standard criteria from the KDOQI CKD Guidelines1: stage 1, persistent albuminuria with glomerular filtration rate (GFR) >90 mL/min per 1.73 m2; stage 2, persistent albuminuria with GFR of 60 to 89 mL/min per 1.73 m2; stage 3, GFR of 30 to 59 mL/min per 1.73 m2; stage 4, GFR of 15 to 29 mL/min per 1.73 m2.
CKD, chronic kidney disease; CI, confidence interval; NHANES, National Health and Nutrition Examination Surveys; NA, data not included because patients with CKD stage 5 were excluded.
From Coresh J, Selvin E, Stevens LA, et al. Prevalence of chronic kidney disease in the United States. JAMA. 2007;298:2038-2047.
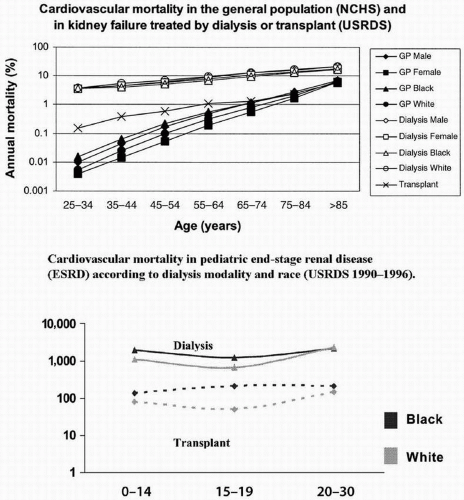
Over the past 10 years, it has been shown that the cardiovascular disease (CVD) burden in patients with kidney disease is enormous. The incidence of CVD among patients with kidney disease is dramatic with an increased risk of mortality from CVD 10 to 100 times greater in patients on dialysis, compared to the general population depending on age (3) (Fig. 11.1A). Younger children are also at increased risk of mortality from CVD and are at up to 1,000 times greater risk compared to the general population, especially infants and children (4) (Fig. 11.1B). Thus, patients with kidney disease regardless of age are at risk, and preventative efforts are needed across all ages. Risk of mortality from CVD after a patient has received a transplant is markedly reduced; however, the risk is still greater than that in the general population.
The incidence of atherosclerotic CVD including fatal and nonfatal events is 146.9 per 1,000 person-years in whites and 118.7 per 1,000 person-years in blacks on dialysis. Incidence of recurrent CVD is 404.1 per 1,000 person-years in whites and 317.5 per 1,000 person-years in blacks on dialysis. Whites are 1.35 (95% confidence interval [CI], 1.18 to 1.55) times more likely to develop incident CVD compared with blacks and 1.25 (95% CI, 1.14 to 1.36) times more likely to develop recurrent disease after adjusting for traditional CVD and dialysis-related risk factors. Along with sudden death, presumably from multifactorial cardiomyopathy, atherosclerotic coronary disease, peripheral vascular disease, and stroke are the most common causes of CVD events occurring among dialysis patients (5,6).
FIGURE 11.1 Cardiovascular mortality in end-stage renal disease (ESRD). A: Cardiovascular mortality in ESRD and the general population (GP) by treatment modality. B: Pediatric cardiovascular mortality in ESRD by treatment modality and race. (Reproduced with permission from Foley RN, Parfrey PS, Sarnak MJ. Clinical epidemiology of cardiovascular disease in chronic renal disease. Am J Kidney Dis. 1998;32 (5 suppl 3):S112-S119. Review. 2) Parekh RS, Gidding SS. Cardiovascular complications in pediatric end-stage renal disease. Pediatr Nephrol. 2005;20:125-131. [Epub 2004 Dec 15].)
Research on modifiable risk factors including dyslipidemia in kidney disease has not found consistent relationships with traditional risk factors and CVD outcome, as kidney disease itself confounds that relationship due to altered metabolism, renal clearance, and also uremia or acidosis, which affects normal cellular function. Dyslipidemias, in fact, are extremely common in CKD; the prevalence among adults ranges from 35% to 85% (based on either elevated low-density lipoprotein cholesterol [LDL-C] or decreased high-density lipoprotein cholesterol [HDL-C], depending on the type of kidney disease (7). This range in the prevalence of dyslipidemia appears similar even among children with CKD. The association with CVD is not a strong linear relationship as in the general population. In fact, total cholesterol (TC) and non-HDL-C demonstrate a positive linear relationship only in those patients on dialysis who do not have any evidence of inflammation. In those with inflammation, the relationship of TC and non-HDL-C and CVD is more complicated (8).
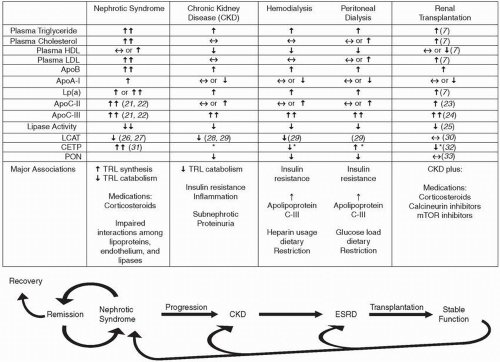
Studies of dyslipidemia in CKD generally divide patients into clinical subsets. It is certainly true that there are clear clinical diagnoses in which a patient demonstrates one type of renal disease to a much greater extent than others. Minimal change nephrotic syndrome, ESRD in an anephric patient, and a highfunctioning kidney transplant are examples of well-delineated clinical renal diseases. For the purposes of this discussion, we define CKD or chronic renal insufficiency as the clinical situation associated with significant loss of GFR and without overt nephrotic syndrome (heavy proteinuria). In discussing nephrotic syndrome, it is implied that patients have only mild to moderate decreases in GFR. Finally, renal transplantation should imply a well-functioning graft. Nonetheless, such divisions are, in practice, incomplete and somewhat inadequate. For example, patients with glomerulonephritis or nephrotic syndrome generally have both heavy proteinuria and decreased renal function. Most, if not all, patients with decreased GFR have moderate, though subnephrotic, proteinuria. Patients undergoing dialysis usually have residual renal function, even to the point of having significant proteinuria. Following transplantation, GFR is not normal. In addition, as indicated in Figure 11.2, an individual patient may pass through several of these scenarios. Thus, it is clear that the elements of dyslipidemia discussed separately below must be considered together in many individual cases.
FIGURE 11.2 Dyslipidemias across the spectrum of kidney disease. (Adapted from Saland JM, Ginsberg HN. Lipoprotein metabolism in chronic renal insufficiency. Pediatr Nephrol. 2007;22:1095-1112 and Saland JM, Ginsberg H, Fisher EA. Dyslipidemia in pediatric renal disease: epidemiology, pathophysiology, and management. Curr Opin Pediatr. 2002;14:197-204.)
The objectives of the chapter are to address the current state of knowledge of the underlying metabolism of lipids in kidney disease, the prevalence of dyslipidemias in kidney disease, and also the latest evidence-based treatment trials in kidney disease.
THEORETICAL CONSIDERATIONS
Dyslipidemias Associated with CKD
Figure 11.2 presents the main lipid and lipoprotein findings in CKD. Individuals with CKD manifest hypertriglyceridemia, usually to a moderate degree. Patients with moderate or severe decline in GFR usually demonstrate normal or borderline high TC and LDL-C, but HDL-C is usually decreased. The characteristic alterations of the major lipoprotein fractions caused by CKD can be summarized as the dyslipidemic triad, that is, increased triglycerides (TG) and very low-density lipoproteins (VLDL); decreased HDL; and increased numbers of small, dense LDL particles. A similar profile is present in patients with diabetes (Chapter 10) and those with the VLDL overproduction syndromes (Chapters 7 and 8). Atherogenic VLDL remnants and chylomicron remnants are also present. Similar lipid and lipoprotein changes have also been noted in studies of children with CKD (9, 11), a finding that strengthens the association of dyslipidemia with renal disease itself, since secondary etiologies of renal insufficiency (such as diabetes) and coprevalent CVD are extremely uncommon in this age group.
Postprandial clearance of TG in CKD is reduced, with both prolonged and greater concentration. Impaired catabolism of chylomicron remnants as well as VLDL remnants competing for lipolysis by lipoprotein lipase (LPL) and apolipoprotein C-II accounts for some of this finding (see also Chapters 7 and 10). Also, intestinal fat absorption itself may be delayed or diminished in severe CKD. With advancing degrees of CKD, the characteristic dyslipidemia becomes more prevalent and individually more pronounced.
The two modalities of chronic dialysis superimpose somewhat different effects upon the background of CKD. In particular, chronic peritoneal dialysis is associated with a relative hypercholesterolemic effect compared to chronic hemodialysis. Major differences between the modalities, such as the large load of glucose for peritoneal dialysis, the use of heparin in hemodialysis, different diet allowances, or differences in the frequency of cyclic variations in fluid volumes or clearance of toxins, are all likely to have metabolic effects. It should be noted, however, that such findings are largely observational, and it is possible also that there are underlying differences between the populations undergoing peritoneal dialysis versus hemodialysis.
Pathophysiologic Mechanisms
Figure 11.2 presents a variety of characteristic findings of CKD on apolipoproteins, enzymes, and regulatory molecules affecting lipoprotein metabolism. The most clear pathophysiologic mechanism leading to increased triglyceride-rich lipoproteins in CKD is that of decreased LPL and hepatic lipase (HL) activity. Indeed, the majority of kinetic studies of lipoprotein turnover in individuals with CKD have demonstrated decreased catabolism, with some also demonstrating an increased VLDL synthetic rate. An important association is the consistent finding of markedly increased apolipoprotein C-III (apoC-III) in the setting of renal insufficiency. ApoC-III is an inhibitor of LPL activity. While elevated TG in humans are generally associated with increased apoC-III levels, an active, rather than a passive, role seems likely. Indeed, apoC-III appears to increase plasma TG by various mechanisms, including modulating the binding of TG-rich lipoproteins to hepatic cell receptors and proteoglycans and directly decreasing the lipolysis of TG by LPL (see also Chapter 7). Whereas transgenic mice expressing excess human apoC-III develop hypertriglyceridemia, apoC-III knockout mice have enhanced ability to catabolize TG. Rare cases of apoC-III deficiency in humans produced similar findings.
The development of decreased HDL and increased concentrations of small, dense LDL may result from metabolic abnormalities that accompany hypertriglyceridemia. With an increased number of triglyceride-rich apolipoprotein B-100 (apoB-100)-containing lipoproteins in circulation, there is an increased transfer of TG from VLDL in exchange for cholesteryl esters (CE) on LDL and HDL. This exchange is due to the enhanced activity of cholesterol ester transfer protein (CETP). As the TG in the cholesterol-depleted LDL and HDL is hydrolyzed by LPL and HL, small, dense LDL and HDL particles are produced (see also Chapters 7, 8, and 10). Whether renal insufficiency affects CETP activity itself remains unclear due to mixed results from a few clinical studies. The decreased lipolysis of VLDL TG due to decreased LPL activity and increased apoC-III in CRD may prolong the half-life of VLDL, making more substrate available for CETP. Finally, with an increased transfer of CE from LDL and HDL to VLDL in exchange for TG, VLDL and its remnants become more cholesterol enriched, rendering them more atherogenic.
The circulating pool of HDL is also markedly affected by increased interactions with triglyceride-rich apoB-100 lipoproteins as mediated by CETP. As HDL become relatively enriched in TG themselves, these particles become more favorable targets of lipases, particularly HL. As HDL are undergoing loss of core TG, the HDL particles shed free apolipoprotein A-I (apoA-I), the major apolipoprotein of HDL. Free apoA-I is filtered by the glomerulus and, via interactions with the megalin/ cubulin complex, is taken up by the proximal tubular epithelium where apoA-I is completely degraded. With loss of GFR as occurs in CKD, this effect may be somewhat blunted, but observations suggest that this pathway is not so limited that HDL or apoA-I levels are preserved. Indeed, when total apoA-I levels among individuals with CKD are very low, circulating levels of free apoA-I are very high, consistent with either increased appearance resulting from more lipolysis of triglyceride-enriched HDL, decreased renal clearance, or both.
These metabolic mechanisms leading to decreased HDL may be exacerbated by abnormalities in the formation and maintenance of HDL. There is some clinical data, as well as support from animal studies, that indicates a deficit in lecith-incholesterol acyltransferase (LCAT) activity in CKD. Substantial HDL maturation requires LCAT activity to esterify free cholesterol garnered from cell membranes, allowing growth of the neutral lipid core of the lipoprotein.
The effects of chronic renal disease also alter the structure and function of lipoproteins via oxidative stress, chronic inflammation, and other toxic components of uremia (10). For example, HDL from uremic patients are frequently contaminated by serum amyloid A. Paraoxonase (PON) is an esterase normally associated with HDL that confers antioxidant activity, but this activity is decreased in CKD. This will promote a “dysfunctional” HDL that will be less efficient in reverse cholesterol transport from peripheral cells such as macrophages back to the liver for excretion in bile (see also Chapter 9). Another example of abnormal lipoprotein structure is sialylation of apolipoproteins, including apoC-III. Given their greater susceptibility to oxidation, the previously mentioned increased proportion of small, dense LDL, as well as the well-known increase in oxidative stress in the setting of CKD, it is not surprising that increased concentrations of oxidized LDL are also encountered.
Only gold members can continue reading. Log In or Register to continue
 Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
 Dyslipidemia in Children and Adolescents
Dyslipidemia in Children and Adolescents
 Dietary Treatment of Dyslipidemia
Dietary Treatment of Dyslipidemia
 Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
 Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
 Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease
Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease



