James S. McCarthy, Thomas A. Moore
Drugs for Helminths
Although a large number of drugs have been used for anthelmintic chemotherapy, treatment is dominated by just three drugs, namely, albendazole, ivermectin, and praziquantel. This is because of their generally high-level efficacy, relatively low cost, and good safety profiles. The spectrum of activity of these drugs can be classified according to the class of helminths against which the drugs have specific efficacy: nematodes or roundworms, for which albendazole or ivermectin are generally used; flukes or trematodes, for which praziquantel is generally used; and cestodes, for which intestinal infection is generally treated with praziquantel and tissue infection with albendazole. Drug doses are given in Table 42-1.
TABLE 42-1
Drugs for Helminth Infections
| DRUG | DOSE | COMMENT |
| Albendazole | 400 mg* | Taken with a fatty meal |
| Mebendazole | 100 mg twice daily for 3 days | Less active than albendazole against extraintestinal infections |
| Triclabendazole | 10 mg/kg once or twice | Fasciola hepatica infections |
| Pyrantel pamoate | 11 mg/kg base × 1 dose; max. dose, 1g | |
| Ivermectin | 150-200 µg/kg once daily × 1-2 days | |
| Diethylcarbamazine | 6 mg/kg/day × 12 days in 3 divided doses | |
| Praziquantel | 40/60 mg/kg/day in 1 or 2 doses × 1 day | 40 mg/kg for Schistosoma haematobium; 60 mg/kg for Schistosoma mansoni and Schistosoma japonicum |
* Dose varies for indication: once for ascariasis and pinworm; once daily for 3 days for hookworm and whipworm; twice daily for 7 days for strongyloidiasis, and 400 mg twice daily for 8-30 days for neurocysticercosis, and for 1-6 months for echinococcosis.
Benzimidazoles
Since the identification in 1961 of the potent antiparasitic activity of thiabendazole, this class of drugs has played a critical role in the treatment of parasitic infections worldwide. The structure of all members of this class is based on a bicyclic ring structure where benzene and imidazole rings are fused. The separate discoveries that thiabendazole is deactivated by hydroxylation of the benzene ring and that activity is enhanced by adding a 2-methylcarbamate moiety to the imidazole ring led to the development of mebendazole and albendazole.
The principal antiparasitic effect of the benzimidazoles (with the exception of triclabendazole) appears to be through selective binding to nematode tubulin, thus preventing polymerization into microtubules, resulting in disruption of cell division and energy pathways,1,2 and interference with similar vital processes, resulting in parasite death. This effect on tubulin also prevents hatching in helminth eggs.3
With the use of albendazole in mass treatment programs, there are concerns about the continued efficacy of the benzimidazoles to treat human infections, particularly given the well-documented reports of resistance from the veterinary literature. Resistance to albendazole in veterinary nematodes is caused by a small number of amino-acid substitutions in the parasite’s β-tubulin protein, the most important of which is a phenylalanine to tyrosine substitution at position 200, with less important changes at residues at 167 and 198.4 Until recently, these concerns were unsubstantiated by evidence of resistance in human isolates. However, recent reports suggest that benzimidazole resistance may be developing in Wuchereria bancrofti5, Trichuris trichiura,4,5a and hookworms,5a,5b particularly in areas where benzimidazoles have been extensively used in lymphatic filariasis control programs. These findings portend potential threats to the future utility of benzimidazoles for treatment of helminth infections.
Albendazole
Albendazole (Fig. 42-1) has a mode of action similar to other benzimidazoles; it is effective against a wide range of helminths and some protozoa. It is directly active against intestinal parasites; thus, the relatively poor absorption of the parent drug from the intestine is ideal for an intraluminal effect. For tissue-dwelling helminths, however, albendazole acts as a prodrug, with the metabolite albendazole sulfoxide being responsible for anthelmintic activity outside the intestinal lumen. The efficacy of albendazole against tissue-dwelling helminth infections, such as echinococcosis, is difficult to reliably predict,6 in part because of variable intestinal levels of the active metabolite in blood and tissues.7
Albendazole is administered orally, either as tablets (200 mg or 400 mg) or as a suspension (2% or 4%), with the dose regimen dependent on the target parasite. Tablets may be swallowed whole, chewed, or crushed and mixed with food. The drug is poorly soluble in water, with alcohol increasing solubility. However, no studies have been undertaken in humans to investigate bioavailability and efficacy when the drug is administered with alcohol.
Bioavailability studies of the parent drug have not been possible because of the lack of availability of a parenteral form. An estimated 5% to 10% of the drug is absorbed after oral administration. Once absorbed, the drug undergoes nearly complete first-pass metabolism either in the gut mucosa or in the liver to the active metabolite albendazole sulfoxide, a mixture of R(+) and S(−) enantiomers. Formation of the R(+) enantiomer of albendazole sulfoxide is catalyzed by microsomal flavin monooxidase, and the S(−) enantiomer, by the cytochrome P-450 enzyme CYP3A8 and in gut epithelium.9 Albendazole sulfoxide is then subject to biotransformation by cytochrome P-450 enzymes (primarily CYP3A4)10 into the inactive metabolite albendazole sulfone.11 The proportion of enantiomer production is species dependent, but in humans, the R(+) enantiomer predominates.12 In patients with neurocysticercosis, albendazole R(+) sulfoxide accumulates in the cerebrospinal fluid (CSF) at a higher level than the S(−) enantiomer.13 Data from a recent study indicated that the R(+) enantiomer is more active than the S(−) enantiomer against Taenia solium.14
The pharmacokinetic profile of albendazole differs between men and women. For the main metabolites, albendazole sulfoxide and albendazole sulfone, there is no significant difference in half-life, time to reach peak concentration (tmax), and mean residence time (MRT). However, the apparent oral clearance (Clp/F) and apparent distribution volume (Vd/F) are lower in women, and the serum peak concentration (Cmax), serum concentration-time curve (area under curve [AUC]), and area under the first-moment curve (AUMC) are lower in men.15
Food enhances the oral bioavailability of albendazole, presumably by stimulating gastric acid secretion, because albendazole absorption is pH dependent. This is true in both healthy patients16,17 and subjects infected with tissue cestodes.18,19 Plasma concentrations of albendazole sulfoxide are up to fivefold higher when albendazole is administered with a fatty meal (fat content ≈ 40 g) compared with the fasting state. The administration of a single 10-mg/kg oral dose of albendazole with a high-fat meal (57 g fat, 1399 kcal) has been shown to increase the mean Cmax and AUC by 6.5- and 9.4-fold, respectively.20 The time to reach Cmax increased from 2.5 to 5.3 hours, compared with the fasting state and administration with water. The elimination half-life was not affected.
Tissue and blood concentrations of albendazole are also determined by mucosal cytochrome P-450 enzymes, which metabolize the drug, and by P-glycoprotein, which acts as an efflux pump into the intestinal lumen.21
In studies of patients with Echinococcus granulosus infection, albendazole administered orally in a dose of 10 to 14 mg/kg/day resulted in stable plasma concentrations of albendazole sulfoxide after 2 to 4 days of treatment. Significant quantities of this metabolite are measurable in lung and liver tissues and in hydatid cyst fluid obtained at surgery.22,23 This active metabolite is excreted in bile. Albendazole sulfoxide crosses the blood-brain barrier to achieve levels in the CSF to approximately 43% of plasma levels.24 However, there is significant interindividual variation because of interindividual differences in drug handling that are influenced by age, gender, or inflammation in the subarachnoid space. The high efficacy of albendazole for treatment of neurocysticercosis is most likely due to penetration of the central nervous system (CNS) by albendazole sulfoxide, which reaches higher brain levels than that achieved in plasma.13
Albendazole is generally taken as a single dose of 400 mg for mass drug administration for intestinal nematode infections in adults and children older than 2 years. However, apart from ascariasis, single-dose therapy, although reducing infection intensity, is not curative. The drug has not been fully evaluated in infants, but in one study of children aged 9 to 23 months, no adverse laboratory abnormalities were noted.25 Because it is mostly metabolized by the liver, albendazole requires no dose adjustments in renal failure. Conversely, absorption of albendazole and clearance of albendazole sulfoxide have been shown to be delayed in patients with echinococcosis and significant biliary obstruction. However, a paucity of data limit dosing recommendations in this group.26 The maximum recommended dosage in humans, regardless of total body weight, is 800 mg/day.
In humans, single-dose albendazole therapy is very well tolerated, with an overall frequency of side effects attributable to the drug to be less than 1%.27 In a large placebo-controlled study involving 700 patients, the incidence of side effects in both groups was equivalent.28 Of importance, it is difficult to reliably differentiate symptoms attributable to the drug itself from the immune response to antigen released from dead parasites. In an analysis of clinical trials where patients with lymphatic filariasis were administered albendazole, or given in combination with other agents, side effects were almost exclusively limited to patients with microfilaremia.27
Liver function abnormalities and bone marrow toxicity have been observed during the prolonged courses of therapy given for treatment of echinococcosis.27 These observations led to the initial recommendation that the drug be administered in treatment cycles of 28 days on/14 days off when prolonged use is anticipated. With increased experience, however, there is less concern about continuous treatment, but monitoring is recommended.29 The most commonly encountered side effects have been transient liver function abnormalities (≤20%) and alopecia (5%).30 Bone marrow toxicity is rarely observed but can be irreversible. Abnormalities in liver function tests are typically less than five times the upper limit of normal and generally return to normal without stopping treatment.
Animal studies have demonstrated that both albendazole and albendazole sulfoxide are teratogenic in rats and rabbits at doses greater than 6 mg/kg/day and 30 mg/kg/day, respectively.32 These and other observations have led to the recommendation that albendazole not be administered during pregnancy. However, reports from clinical trials where single-dose albendazole therapy has been administered to pregnant women with hookworm infection (treated after the first trimester) demonstrated no effect on perinatal mortality, congenital malformations, or birth weight.33 Although a small reduction in human immunodeficiency virus (HIV) viral load was noted,34 no effect on vertical transmission of HIV was observed.35 Of note, the trials demonstrated an increase in childhood eczema among treated children.33,36
Cimetidine inhibits the absorption of albendazole through reduction of gastric acidity, yet inhibits metabolism of albendazole sulfoxide by interfering with CYP3A4 enzymes, thus prolonging the elimination half-life from 7.4 (+/−3.3) hours to 19.0 (+/−11.7) hours.37 Grapefruit juice inhibits metabolism of albendazole at the intestinal mucosa, but drug concentrations are higher than among those administered cimetidine.20 Short-term administration of ritonavir, a potent CYP3A4 inhibitor, does not significantly alter the pharmacokinetic parameters of albendazole. However, long-term administration results in significant decreases in AUC and Cmax.38 The blood concentration of albendazole sulfoxide is increased by 50% when administered concurrently with dexamethasone.39 This has clinical relevance in the setting of treatment of neurocysticercosis, where albendazole is usually administered in conjunction with corticosteroid cover to prevent reactive cerebral edema caused by parasite death.
Mebendazole
Mebendazole (Fig. 42-2) was first introduced in 1977 as a veterinary anthelmintic agent for treatment of Echinococcus multilocularis. Its mode of action is similar to other benzimidazoles. Although it has been approved for the treatment of both intestinal and tissue helminths, it is less effective than albendazole for treatment of extraintestinal helminths, and therefore, it is used almost exclusively for the treatment of common intestinal nematode infections.
Like albendazole, mebendazole is poorly soluble, poorly absorbed, and undergoes extensive first-pass metabolism in the liver. However, it is even less well absorbed than albendazole, with a bioavailability of only 1% to 2% after administration of a single oral dose. The low bioavailability is attributable both to the low solubility of the oral formulation and to the high-level of first-pass metabolism in the liver. Ingestion with fatty food increases absorption. The drug is highly protein bound (≈95%). The absorbed portion of mebendazole is predominantly metabolized by the liver. There are two major metabolites: 2-amino-5-benzoylbenzimidazole, created by amide hydrolysis, and methyl-5[α-hydroxybenzyl]-2-benzimidazole carbamate, a product of ketone reduction. Unlike albendazole, these and other metabolites are not believed to have significant anthelmintic activity.
Mebendazole reaches its highest tissue concentrations in the liver. About half of the absorbed dose is excreted in the urine as metabolites40; however, a significant portion is also excreted in bile as metabolites. Mebendazole crosses the blood-brain barrier but reaches levels significantly lower than serum.
Cimetidine appears to improve the bioavailability of mebendazole.41 Among patients with cystic echinococcosis, there is significant variability in absorption. Furthermore, concomitant administration of phenytoin and carbamazepine results in lower plasma levels, presumably because of induction of the cytochrome P-450 enzyme CYP3A4. Although no other significant drug-drug interactions have been reported with mebendazole, like albendazole, caution is warranted among individuals prescribed prolonged treatment courses and who are also taking medications with effects on the P-450 system.38
For the treatment of soil-transmitted helminths, mebendazole is typically administered as 100 mg given twice daily for 3 days. However, its pharmacokinetic profile enables a single 500-mg dose to be administered for mass treatment campaigns to control soil-transmitted helminths. Although mebendazole has not been fully evaluated in children 2 years or younger, it is well tolerated in community geohelminth control programs.42,43 The maximum recommended dosage in humans, regardless of total body weight, is 500 mg/day.
Although metabolites are excreted in the urine, there are no clinical data on the use of mebendazole in patients with kidney disease, and dose adjustment does not appear to be necessary in this setting. Nevertheless, caution seems warranted in these patients; therefore, if a prolonged course of therapy is anticipated, drug levels (if available) should be obtained. Similarly, there are no clinical data on the use of mebendazole in patients with liver disease, but increased drug levels have been observed in a patient with cholelithiasis.44
When given either as a single 500-mg dose or 100 mg twice daily for 3 days, mebendazole is very well tolerated. In a recent trial involving more than 600 children given treatment for a geohelminth eradication program in Zanzibar, the most common adverse events were limited to abdominal cramps (11%), fatigue (6%), headache (6%), vertigo (4.4%), and nausea (3.7%).45 This is similar to reports from other large clinical trials. When taken at higher doses for longer periods (50 mg/kg for 3 to 12 months) for the treatment of echinococcosis, side effects occurred in approximately 20% of patients, were minimal and reversible in all patients without discontinuing treatment (transient elevation of transaminases, abdominal pain, headache, vertigo, urticaria, and dyspepsia).46,47 There are also case reports of reversible bone marrow suppression.48
Although teratogenic in rats, the safety of mebendazole in pregnancy has been examined in three large human studies. In a survey of 170 women who took mebendazole in the first trimester of pregnancy, fetal loss or neonatal death was not significantly higher than that observed in the general population.49 In a second series, one congenital hand malformation was observed in 112 first-trimester exposures.49 In a larger retrospective study sponsored by the World Health Organization (WHO), involving more than 7000 Sri Lankan women who had accidentally taken the drug anytime during their pregnancy, there was a significantly lower rate of fetal loss and perinatal death, presumably resulting from reduced levels of maternal anemia.50 Despite this ostensible beneficial effect, there was a trend toward a higher rate of congenital malformations among those who were exposed during the first trimester (2.5% vs. 1.5%). In a randomized, placebo-controlled trial in Peru, where women in the second and third trimesters were enrolled, no differences in the incidence of adverse effects were observed between the treatment arm compared with those receiving placebo.51 The beneficial effect of decreased incidence of very low birth weight was again noted.
Thiabendazole
Thiabendazole (Fig. 42-3) remains one of the most potent of the benzimidazoles developed; however, its use has dramatically declined, and its availability has become increasingly limited because of the higher frequency of side effects compared with other equally effective agents from this class. It is active against most intestinal nematodes that infect humans and is active against the intestinal phase of Trichinella spiralis infection in humans. Although widespread resistance of helminths to thiabendazole has been reported in veterinary literature, there have been no reports of resistance in humans.
Although the mechanism of anthelmintic activity of thiabendazole remains unknown, it is presumed to be similar to the other benzimidazoles. Unlike mebendazole, thiabendazole has been shown to specifically inhibit fumarate reductase, a helminth-specific enzyme in the muscle-stage larvae of T. spiralis.52 In addition, animal studies have shown that thiabendazole exhibits anti-inflammatory, analgesic, and antipyretic effects, which may explain its putative benefit in patients suffering from trichinosis and dracunculiasis.
Thiabendazole is rapidly absorbed from the gut and can also be absorbed through the skin. After administration of a single oral dose, plasma levels peak within 1 to 2 hours, and most of the drug is cleared from the plasma within 8 hours.53 The drug should be taken after meals, primarily to reduce gastrointestinal (GI) side effects, which are common. Thiabendazole is extensively metabolized in the liver to 5-hydroxythiabendazole before being excreted as glucuronide or sulfate conjugates. Most of the drug metabolites are excreted within the first 24 hours; at 48 hours, 87% of a single oral dose of thiabendazole is excreted in urine, and 5% is excreted in feces.
The most commonly reported adverse effects include anorexia, nausea, vomiting, diarrhea, headache, and dizziness. Neurologic side effects can be significant and include drowsiness, visual disturbances, and giddiness. Therefore, tasks requiring mental acuity should be avoided during treatment. Stevens-Johnson syndrome and angioneurotic edema have been reported, albeit rarely. Patients notice an asparagus-like odor of the urine that resolves within 24 hours of discontinuation of treatment.
Thiabendazole dosing is determined by actual weight, but some treatment regimens are parasite specific. Data regarding the safety and efficacy in children weighing less than 10 kg are not available. No specific adjustments are recommended in patients with renal or hepatic failure; only cautious use is advised. Thiabendazole has not been shown to cause birth defects or other problems in studies in rabbits, rats, and mice given 2.5 to 15 times the usual human dose. However, cleft palate and bone defects were observed in mice given 10 times the usual human dose.54 It is unclear if thiabendazole is significantly excreted into breast milk.
Thiabendazole is a potent inhibitor of CYP1A2.55 Thus, caution is warranted in patients also taking caffeine, theophylline, or melatonin because levels of these medications can be increased.
Triclabendazole
Triclabendazole (Fig. 42-4) is a benzimidazole compound used routinely since 1983 in veterinary practice for the treatment of fascioliasis. It was first used for the treatment of human infection in 1986; however, its use in humans expanded in 1989 during an outbreak of fascioliasis near the Caspian Sea, when Iranian authorities approved the use of the veterinary formulation to treat human infections. In 1997, after the remarkable success of clinical trials using triclabendazole for the treatment of fascioliasis and paragonimiasis, the WHO recommended that the drug be placed on the Essential Drugs List. Although the U.S. Food and Drug Administration (FDA) has not approved triclabendazole for use in humans, it is registered in some countries where fascioliasis is endemic.
Triclabendazole is a narrow-spectrum agent and is unique among benzimidazoles in its highly specific activity against Fasciola spp. and Paragonimus spp., with minimal activity against nematodes, cestodes, or other trematodes. Like other benzimidazoles, the mechanism of action of triclabendazole results from inhibition of microtubule formation.56 The β-tubulin protein of F. hepatica exhibits species-specific protein changes at position 82 (glutamic acid) and position 91 (threonine), which are unique among nematodes and cestodes. These substitutions appear to cause the fluke β-tubulin to adopt a three-dimensional structure that is relatively unaccommodating to other benzimidazoles, which are flat or L-shaped.57 Triclabendazole exhibits a nonplanar U-shaped configuration that appears to be uniquely suited to binding to fluke β-tubulin.56 In addition, triclabendazole-sulfoxide, the active metabolite of triclabendazole, has been shown to disrupt the tegument of both mature and immature stages of F. hepatica.58 Furthermore, it is also a potent inhibitor of protein synthesis.59
Although no resistance to triclabendazole has yet been reported in humans, resistance in veterinary use has become widespread since its original description in Australia.60 Susceptibility to the drug appears to be enhanced by ketoconazole61 and methimazole62 among strains previously known to be resistant.
After oral ingestion, absorption of triclabendazole occurs rapidly.63 Like albendazole and mebendazole, triclabendazole undergoes extensive first-pass metabolism in the liver. It is converted into the active metabolite triclabendazole-sulfoxide, and then the inactive metabolite triclabendazole-sulfone.64 Both metabolites are highly protein bound (>99%).63
After oral administration, the parent drug cannot be detected in plasma. Food enhances absorption of triclabendazole but also shortens the elimination half-life of its metabolites.64
Although there are no clinical data regarding treatment of patients with renal disease, dose adjustment seems unnecessary in renal disease, given the short course of therapy and the drug’s extensive hepatic metabolism. Although dose adjustment may be necessary in patients with hepatic disease, no data exist to provide insight.
Triclabendazole appears to be remarkably safe. Adverse events, when they have occurred, have been mild, short lived, and limited to abdominal pain, headache, nausea, and fatigue.65 Abdominal pain, lasting less than 5 days, was reported by 21.5% of patients receiving triclabendazole, 5 mg/kg daily for 3 days, 6.7% receiving 10 mg/kg twice daily for one day, and 31.3% receiving a single dose of 10 mg/kg.66 A recently concluded trial in the Bolivian Altiplano reported similar findings.65 The abdominal pain is typically located in the right upper quadrant, is relieved by oral spasmolytics, and has been attributed to the expulsion of dead or dying worms from the hepatobiliary system into the intestinal tract.64,67,68 The contention that nearly all the adverse reactions in human clinical studies can be attributed to the death of adult worms is supported by ultrasound studies that have demonstrated dilated intrahepatic bile ducts caused by transient biliary obstruction associated with expulsion of dying worms. This is further supported by evidence from clinical trials in paragonimiasis, where the only GI side effects reported were rare episodes of diarrhea. Fever has been reported in 6.3% of patients within 4 days of treatment for paragonimiasis. There have been no reports of derangement of liver function tests, renal function, or hematologic indices attributable to triclabendazole in human clinical trials. However, laboratory studies using high dosages of the drug in rats and dogs have demonstrated bone marrow depression and increased serum alkaline phosphatase. No evidence of dose-related toxicity or carcinogenicity has been observed in animals. Unlike the other benzimidazoles, triclabendazole has not been shown to cause birth defects in animal studies. Nevertheless, there are no data regarding its safety in pregnancy. Where possible, it seems prudent to avoid use in the first trimester. Triclabendazole is known to pass into breast milk, but there are no reports of adverse events in nursing babies. No data exist regarding drug-drug interactions.
Ivermectin
Ivermectin is a semisynthetic antibiotic derived from Streptomyces avermitilis. The organism was isolated from a soil sample taken from a golf course in Japan. Ivermectin is marketed under the brand names Stromectol (Merck, Whitehouse Station, NJ) in the United States, Mectizan (Merck) in Canada, and Ivexterm (Valeant Pharmaceuticals, Montreal, Canada) in Mexico. It is lipophilic, with poor water solubility. It is a mixture of approximately 80% molecular structure C48H74O14 (22,23-dihydroavermectin B1a) and approximately 20% molecular structure C47H72O14 (22,23-dihydroavermectin B1b), with an average molecular weight of 1736.2 (Fig. 42-5). The relative proportion of the B1a and B1b isoforms is a result of the fermentation process; both have nearly identical antiparasitic activity. After the discovery of its activity against nematodes of veterinary importance, it was later found to be extremely effective against the cattle parasite Onchocerca cervicalis; this led to its development for treatment of onchocerciasis.69 It is active at low doses against a wide range of helminths and ectoparasites and is the drug of choice for treatment of onchocerciasis and strongyloidiasis. It is an option for treatment of cutaneous larva migrans, head lice, and scabies. Although active against the intestinal helminths Ascaris lumbricoides and Enterobius vermicularis, it is only moderately effective in trichuriasis when given alone and has limited activity against hookworms.
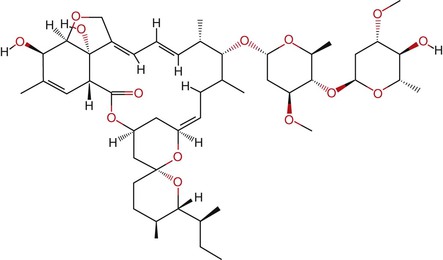
Ivermectin activates neuromuscular membrane-associated, chloride channels, particularly glutamate-gated channels, by binding to α-type channel subunits. The subsequent influx of chloride ions results in hyperpolarization and muscle paralysis, particularly of the nematode pharynx, thus halting ingestion of nutrients. Ivermectin binding sites have been identified in rat brain, but the affinity of the drug for nematode ligands is about 100 times greater than for mammals, likely explaining its selectivity.
It is available for human use only as an oral formulation (either a 3-mg tablet or 6-mg scored tablet) and is generally administered as a single dose of 150 to 200 µg/kg. The bioavailability of ivermectin is increased twofold with food.70,71 Plasma concentrations of ivermectin have been shown to decrease with ingestion of orange juice72 and increase with ingestion of beer.73 Its absorption half-life is approximately 1 hour,74 with Cmax being reached in 4.7 to 5.6 hours.75,76 The Cmax is proportional to dose, with a value of approximately 38 to 46 µg/L reached after a therapeutic 150- to 200-µg/kg dose.75–77 No significant differences in absorption have been found between healthy volunteers and patients with onchocerciasis. A second rise in plasma levels occurs 6 to 12 hours after ingestion, suggesting significant enterohepatic recycling.74 Because it is a highly lipophilic drug, ivermectin is distributed widely throughout the body. Animal studies indicate that it accumulates at the highest concentration in adipose tissue and liver. Plasma protein binding is 93%.78 The terminal half-life of the parent drug is 20 hours.79 The elimination half-life of the metabolites of ivermectin is longer than that of the parent drug, at about 3 days.79 It is not known whether the metabolites have antiparasitic activity. In healthy men, the volume of distribution in the central compartment (Vc) was 3.1 and 3.5 L/kg, after ingesting 6 and 12 mg of ivermectin, respectively.80 In patients with onchocerciasis, the volume of distribution of the area (Vl) was 9.9 L/kg after administration of a single 6-mg tablet.76 The tissue distribution was similar in both groups of patients. A two-compartment model of distribution is consistent with the disposition of ivermectin in humans and other species, with a high volume of distribution into a peripheral compartment.81 Ivermectin is metabolized extensively in the liver via the cytochrome P-450 isozyme CYP3A4.82
Ivermectin is both a substrate for the transporter P-glycoprotein (Pgp),83,84 as well as a moderately potent Pgp inhibitor at concentrations consistent with doses used in mass treatment campaigns.85,86 It is highly protein bound; scant pharmacokinetic/pharmacodynamic data exist regarding levels in individuals with conditions that would affect these parameters. The lack of significant macrofilaricidal activity in onchocerciasis cannot be explained by lack of penetration into onchocercal nodules, as the drug penetrates well into nodules when given as a single dose.75 In an animal model of onchocerciasis, high concentrations were detected in the capsule wall and inside the nodule after subcutaneous administration with 500 µg/kg.87 Because there is no significant renal handling of the drug, administration in renal failure should not be of concern. There are no data regarding safety in hepatic failure, but it is probably safe for use in this setting, given the lack of significant side effects at high doses.70 Less than 1% of the drug is excreted in the urine as 3′-O-demethyl-22,23-dihydroavermectin B1a and 23-dihydroavermectin B1a. The remainder is excreted as metabolites in the feces, mainly as monosaccharide derivatives, which also exhibit enterohepatic recycling.88
In strongyloidiasis, the difficulty in achieving adequate drug levels of ivermectin in patients with disseminated strongyloidiasis and intestinal ileus can be a serious clinical problem. In one patient with disseminated strongyloidiasis, ivermectin levels were below the average of those reported by other authors after oral administration (1.1 ng/mL) when ingesting a total dose of 1000 µg/kg over 3 consecutive days. This was then followed by the subcutaneous administration of a veterinary formulation of ivermectin at a dose of 200 µg/kg given every 2 days, which resulted in ivermectin levels to 7.9 ng/mL 1 week after the last dose, with evidence of additional metabolite accumulation and a sustained antiparasitic effect.89 An additional report has demonstrated improved absorption with the subcutaneous route, using the same veterinary solution.90 The serum ivermectin concentration, drawn 3 hours after the third oral daily dose, was only 0.8 ng/mL. However, it increased to 5.8 ng/mL 16 hours after the first subcutaneous dose. Serum levels of 11.4 to 17.2 ng/mL were noted over the next 15 days, without evidence of significant accumulation.
Pgp appears to be important in preserving the blood-brain barrier, and preventing accumulation of ivermectin in mammalian brain tissue.91 Pgp is an essential component of the intact blood-brain barrier in vivo. In knockout mice deficient for Pgp, the 50% lethal dose (LD50) of ivermectin is 100-fold lower than in wild-type mice.83 Collies and some other breeds of dogs are unusually susceptible to the toxic effects of ivermectin, resulting from a mutation in the canine multidrug-resistance gene, designated MDR1-1,92 which results in increased CNS penetration of the drug. Differences in levels of Pgp expression, protein levels, and drug passage across the gut wall and blood-brain barrier have been reported in different age groups, both in laboratory animals and in human studies.93 However, how these results relate to potential CNS toxicity of ivermectin is uncertain, given the interspecies variations and limited data available.94,95 It is unclear whether ivermectin is safe to use in infancy, when expression of Pgp at the blood-brain barrier is likely at its lowest levels; hence, it is recommended not to administer the drug to children weighing less than 15 kg. Although not specifically studied, ivermectin has been used without dosage alteration to treat elderly individuals in mass treatment campaigns. Ivermectin use in the elderly was reported to be associated with an increase in mortality within 6 months after treatment.96 However, this report has been criticized for not matching the patients for comorbidities.
The successful mass distribution programs with ivermectin to control onchocerciasis have prompted the development of programs entailing co-administration of albendazole and azithromycin. The pharmacokinetic interactions of these agents have not been well studied. In a study where azithromycin, ivermectin, and albendazole were co-administered, ivermectin AUC and Cmax were increased by 31% and 27%, respectively.71 A population model developed to analyze drug interaction data suggests the maximum concentration of ivermectin achieved in the interaction phase would not exceed 201 ng/mL.97
Ivermectin is the drug of choice for the treatment of onchocerciasis, called river blindness, which is caused by infection with the filarial nematode Onchocerca volvulus. Onchocerciasis is endemic in Central and Western Africa and until recently in certain areas of Central and South America. Administered as a single oral dose of 150 µg/kg, it has a rapid effect on microfilariae, which cause most of the ocular and cutaneous manifestations of this disease. Most microfilariae disappear by the end of the first week of therapy; within 1 month after treatment with ivermectin, skin microfilarial loads decrease by 95% to 99%.98 The drug also blocks the production of new microfilariae by the adult female worms, which only resume release of microfilariae 3 to 6 months after treatment.99 This embryostatic action of ivermectin explains why the microfilarial burden remains at a very low level for up to 1 year after treatment. Ivermectin also has been reported to exhibit a mild macrofilaricidal effect in reducing the longevity of the adult worms when treatments are repeated every 1 to 3 months.100,101 Together, these effects result in a reduction of morbidity and transmission intensity. The adult worm may live more than 10 years; therefore, repeated doses of ivermectin may be required every 6 to 12 months for the life of the adult worm.
A single dose of ivermectin is similarly microfilaricidal against Wuchereria bancrofti. With a single oral dose of 10 to 20 µg/kg, microfilaremia disappears for up to 3 months.102 With higher doses (150 to 400 µg/kg), microfilaremia takes longer to return, and the level of parasitemia is lower than baseline.103–105 Ivermectin appeared to be slightly inferior to diethylcarbamazine (DEC) in producing sustained reduction of microfilaremia, even when given in combination with albendazole.106 In lymphatic filariasis, as in onchocerciasis, ivermectin has no discernible effect on the adult worm, even when given at a dose of 4800 µg over 6 months.107 Given these data, it is not surprising that, when given as part of a mass treatment campaign, ivermectin resulted in a significantly smaller impact on hydrocele prevalence than DEC.108,109
Although the activity of ivermectin against Brugia malayi and Brugia timori—the less common lymphatic filarial parasites—is less well studied, it appears to be less effective against these species compared with W. bancrofti. Single doses of ivermectin, even as high as 400 µg/kg, result in slower clearance of microfilaremia, and this effect is shorter lived.110 However, this dose does result in a significant reduction in microfilaremic burden, and the level of microfilaremia remains suppressed for up to 2 years, suggesting, as with W. bancrofti, an effect on the adult worms. The addition of albendazole to both DEC and ivermectin treatments does not increase the efficacy of ivermectin against Brugia spp.111
The effect of ivermectin against Loa loa appears similar to that seen in brugian filariasis: high doses of the drug (400 µg/kg) are required to clear microfilaremia112 and the microfilaremia clears more slowly than in bancroftian filariasis.113 Serious adverse events, most notably fatal encephalopathy, have occurred after the administration of ivermectin for the mass treatment of onchocerciasis in areas endemic for loiasis. This led to a temporary suspension of ivermectin distribution in these areas, but this has now resumed after institution of a tiered strategy. Serious adverse effects have been confined to individuals with high levels of microfilaremia (>30,000 microfilariae/mL); ivermectin should therefore be used with extreme caution in these patients.
A single oral dose of ivermectin resulted in sustained suppression of microfilaremia in patients infected with Mansonella streptocerca114 and in a significant reduction of intensity and prevalence of infection in Mansonella ozzardi.115,116 Ivermectin does not appear to be effective against Mansonella perstans.
Ivermectin is the drug of choice for the treatment of infection with Strongyloides stercoralis. Administration of a single 200-µg/kg dose to children with uncomplicated infections resulted in cure rate of 83%.117 This finding is consistent with other clinical data.118–120 To increase the likelihood of cure, most experts recommend an additional dose given 7 to 10 days later. Repeated courses may be needed in individuals with impaired cellular immunity, particularly patients infected with human T-cell lymphotropic virus type 1.121 Although uncomplicated infection can be readily and easily treated with the orally administered drug, patients with disseminated strongyloidiasis (hyperinfection syndrome) present significant challenges. In this condition, paralytic ileus and the frequent need for nasogastric drainage results in poor absorption of ivermectin by enteric routes.89,90 As noted above, the hyperinfection syndrome has been successfully treated by using a veterinary parenteral preparation of ivermectin administered by the subcutaneous route.89,90,122 Ivermectin has also been given by rectal enema.123 However, treatment is not reliably effective, and the pharmacokinetic profile of the drug when administered by means other than the oral route is poorly understood.124
Ivermectin is highly effective for treatment of cutaneous larva migrans (CLM), a zoonosis usually caused by hookworms of dogs (Ancylostoma caninum) or cats (Ancylostoma braziliense). For this indication, it is given at a dose of 150 to 200 µg/kg once daily for one to two doses.125
When given at a dose of 200 µg/kg/day for 2 days, ivermectin is effective for the treatment of gnathostomiasis, with a reported cure rate of 100% in one study.126 When administered as a single dose of 200 µg/kg, cure rates range from 76% to 92%.127 Of those not cured, the signs of resurgent infection (as demonstrated by the recurrence of subcutaneous swelling) were not statistically significant when compared with placebo.128 The drug is reasonably well tolerated in this disease, with self-limited adverse effects that are not dose-related.128
Ivermectin is safe and highly effective against A. lumbricoides. A single dose of 100 to 200 µg/kg results in cure.129 It is also active against pinworm, E. vermicularis, with a cure rate of up to 85% after a single oral dose ranging from 50 to 200 µg/kg.129 However, it has limited activity in hookworm infection, with treatment resulting in reduction of worm burden but not usually cure.129–131
Similar to its activity against hookworm, ivermectin is relatively ineffective for the treatment of infections caused by T. trichiura. The cure rate after a single dose of 50 to 200 µg/kg ranges from 11% to 67%.129,130 Higher doses131 or extending daily treatment to 3 days117 appear to be more effective. In this infection, combination with albendazole appears to result in a greater efficacy than when either agent is used alone.132–134
Although recommended as a second-line alternative to topical permethrin,135 ivermectin is effective for the treatment of infection caused by Sarcoptes scabiei. The recommended dose is 200 µg/kg orally as a single dose, with a second dose given 2 weeks later.135,136 Ivermectin is effective for the more-difficult-to-treat crusted (Norwegian) scabies,137 although repeated treatments are recommended. In this setting, it should be combined with a topical ascaricide, such as permethrin, alternating with keratolytic creams, such as salicylic acid or lactic acid/urea, to facilitate breakdown of the skin crusting.135 A topical formulation of ivermectin 0.4% has recently been approved by the FDA for control of head lice after the report of a randomized, controlled trial where efficacy of 73.8% was reported.138 Of note, however, short exposure intervals of body lice to sublethal amounts of ivermectin was shown to induce upregulation of detoxification genes, including cytochrome P-450 monooxygenase and ATP-binding cassette transporter genes, leading to tolerance.139 This suggested the vulnerability of this class of drugs to the development of resistance.
Ivermectin given in the absence of helminth infection has few if any side effects, as demonstrated by its continued use in mass treatment campaigns for more than 20 years. Although it does not cross the blood-brain barrier in vertebrates, high doses administered in animal studies and observations of human overdose indicate evidence of CNS toxicity, including emesis, mydriasis, and ataxia.140 No significant toxicity was observed after administration of doses as high as 2000 µg/kg, 10 times the recommended therapeutic dose.70,141 However, as noted above, patients with high parasite burden, for example with high levels of microfilariae in the skin (onchocerciasis) or blood (lymphatic filariasis or loiasis) may have significant post-treatment reactions, including postural hypotension, and thus should be observed for up to 36 hours after treatment. In onchocerciasis, skin edema, pruritus, and mild eye irritation may also occur. Usually, recovery follows rapidly when the patient remains recumbent and no specific treatment is necessary. On occasion, patients require symptomatic treatment with antipyretics or antihistamines.
As noted above, severe complications of ivermectin therapy can occur among patients treated for onchocerciasis but with coincident heavy Loa loa infection (>30,000 microfilariae/mL blood). These include encephalopathy, characterized by confusion, urinary incontinence, lethargy, and coma. This reaction led to the temporary suspension of ivermectin distribution in regions where the two filarial infections are co-endemic. A single case of hepatitis associated with ivermectin use has been reported,142 but there are no other reports of significant immune reactions with this agent.
Not surprisingly, the use of ivermectin in mass treatment campaigns for 2 decades has occasionally resulted in the inadvertent administration of ivermectin to pregnant women.143 Although no adverse effects have been recorded in retrospective studies,144,145,146 administration of the drug in pregnancy is not advised. However, the established teratogenicity of the alternative treatments for strongyloidiasis (the benzimidazole drugs thiabendazole or albendazole) suggests that ivermectin would be the drug of choice if life-threatening hyperinfection occurred in a pregnant woman. After a therapeutic dose, ivermectin reaches levels in breast milk that are about 35% of those seen in the serum.147 Although it should be avoided during lactation, operational constraints and the absence of observed toxicity in human studies have resulted in the decision not to exclude lactating women from mass treatment programs.
Because of overuse of ivermectin as a single agent for control of parasites in cattle, resistance to ivermectin has developed in Haemonchus contortus, the organism for which ivermectin was originally developed as a treatment.148 The mechanism of resistance in veterinary nematodes has not been precisely defined and may involve both target alteration and drug pumps. More than 1.3 billion tablets have been distributed in Africa, with some individuals having now received up to 20 annual treatments.149,150 However, in some areas, this program has not resulted in interruption of transmission.151 Reports of poor parasitologic responses to the drug152–154 have been followed by parasitologic and epidemiologic evidence of ivermectin resistance.155 Further, in a study conducted in Cameroon, parasites obtained from individual patients demonstrated changes in the β-tubulin gene before and after those same patients were treated with ivermectin.156 Although these genetic changes developed over the 3 years the study was conducted, these parasites develop slowly, requiring about 1 year to go from birth to sexual maturity.
Related posts:





Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


