Thrombocytopenia is caused by immune reactions elicited by diverse drugs in clinical practice. The activity of the drug-dependent antibodies produces a marked decrease in blood platelets and a risk of serious bleeding. Understanding of the cellular mechanisms that drive drug-induced thrombocytopenia has advanced recently but there is still a need for improved laboratory tests and treatment options. This article provides an overview of the different types of drug-induced thrombocytopenia, discusses potential pathologic mechanisms, and considers diagnostic methods and treatment options.
Key points
- •
Thrombocytopenia is caused by immune reactions elicited by diverse drugs in clinical practice.
- •
The activity of the drug-dependent antibodies produces a marked decrease in blood platelets and a high risk of serious bleeding.
- •
Understanding of the cellular mechanisms that drive drug-induced thrombocytopenia has advanced recently but there is still a need for improved laboratory tests and treatment options.
- •
This article includes an overview of the different types of drug-induced thrombocytopenia, discusses potential pathologic mechanisms, and considers diagnostic methods and treatment options.
History, definition, and classification
Purpura in association with pestilential fevers was described by Hippocrates, but it was not until the sixteenth century that purpura in the absence of fever was observed by Lusitanus. It took another 3 centuries for Krauss (1883) and Denys (1887) to recognize that purpura hemorrhagica was caused by a decrease in blood platelets. The association of drug and purpura was first reported by Vipan in 1865 before the role of platelets in hemostasis was known. Later, Rosenthal in 1928 provided evidence that drug ingestion caused thrombocytopenia by rechallenging a patient with quinine-induced thrombocytopenia with the drug and showing a prompt decrease in platelet count.
Drug-induced thrombocytopenia (DIT) can be divided into 2 categories according to the mechanism responsible for the thrombocytopenia, namely (1) suppression of platelet production and (2) increase in peripheral platelet destruction or clearance. The former, which usually occurs as pancytopenia, is caused mostly by myelosuppressive agents (mainly chemotherapeutic drugs) and their suppressive effect is dose dependent. The latter can be further divided into 3 subtypes:
- 1.
Nonimmune DIT (caused by a direct toxic effect of the drug on platelets; eg, ristocetin, an antibiotic that is no longer used in clinical practice)
- 2.
Immune DIT (mediated by a drug-dependent antibody)
- 3.
Autoimmune DIT (mediated by a drug-independent antibody that binds and prematurely clears platelets even in the absence of the drug such as alpha-methyldopa)
Most drugs are thought to cause thrombocytopenia by a drug-dependent immune mechanism (ie, the second mechanism); this is the focus of this article and other types of DIT are not discussed.
Drug-induced immune thrombocytopenia (DITP) is a common problem in clinical practice, particularly in hospital inpatients. Patients usually develop extensive petechiae or purpura, with markedly low blood platelet levels (frequently <10 × 10 9 /L), about 1 to 2 weeks after commencing a new medication. Serious bleeding including intracranial hemorrhage can occur, presenting a challenging diagnostic and management problem. Diagnosis of DITP is usually made clinically but can be difficult when there are comorbid conditions (eg, infection) and concomitant drugs that can also cause thrombocytopenia. Despite significant advances in this area of hematology, there is still a need for more research to increase understanding of the mechanisms of DITP, and to improve diagnostic tests and patient management. This article discusses the pathogenesis, diagnosis, and management of DITP, focusing on recent advances.
History, definition, and classification
Purpura in association with pestilential fevers was described by Hippocrates, but it was not until the sixteenth century that purpura in the absence of fever was observed by Lusitanus. It took another 3 centuries for Krauss (1883) and Denys (1887) to recognize that purpura hemorrhagica was caused by a decrease in blood platelets. The association of drug and purpura was first reported by Vipan in 1865 before the role of platelets in hemostasis was known. Later, Rosenthal in 1928 provided evidence that drug ingestion caused thrombocytopenia by rechallenging a patient with quinine-induced thrombocytopenia with the drug and showing a prompt decrease in platelet count.
Drug-induced thrombocytopenia (DIT) can be divided into 2 categories according to the mechanism responsible for the thrombocytopenia, namely (1) suppression of platelet production and (2) increase in peripheral platelet destruction or clearance. The former, which usually occurs as pancytopenia, is caused mostly by myelosuppressive agents (mainly chemotherapeutic drugs) and their suppressive effect is dose dependent. The latter can be further divided into 3 subtypes:
- 1.
Nonimmune DIT (caused by a direct toxic effect of the drug on platelets; eg, ristocetin, an antibiotic that is no longer used in clinical practice)
- 2.
Immune DIT (mediated by a drug-dependent antibody)
- 3.
Autoimmune DIT (mediated by a drug-independent antibody that binds and prematurely clears platelets even in the absence of the drug such as alpha-methyldopa)
Most drugs are thought to cause thrombocytopenia by a drug-dependent immune mechanism (ie, the second mechanism); this is the focus of this article and other types of DIT are not discussed.
Drug-induced immune thrombocytopenia (DITP) is a common problem in clinical practice, particularly in hospital inpatients. Patients usually develop extensive petechiae or purpura, with markedly low blood platelet levels (frequently <10 × 10 9 /L), about 1 to 2 weeks after commencing a new medication. Serious bleeding including intracranial hemorrhage can occur, presenting a challenging diagnostic and management problem. Diagnosis of DITP is usually made clinically but can be difficult when there are comorbid conditions (eg, infection) and concomitant drugs that can also cause thrombocytopenia. Despite significant advances in this area of hematology, there is still a need for more research to increase understanding of the mechanisms of DITP, and to improve diagnostic tests and patient management. This article discusses the pathogenesis, diagnosis, and management of DITP, focusing on recent advances.
Causative drugs
More than 200 drugs (including some herbal medicines) have been reported to cause DITP. Aster and Bougie identified 85 drugs that they considered as definite or probable causes of thrombocytopenia. Among these are drugs commonly used in clinical practice: heparin, quinine, antibiotics (eg, penicillin, cephalosporin, rifampicin, vancomycin, sulfonamides), antiplatelet agents, glycoprotein IIb/IIIa (GPIIb/IIIa) inhibitors (tirofiban, eptifibatide, abciximab), antirheumatic agents (gold, d -penicillamine), antiepileptics (sodium valproate, phenytoin), cardiac agents (amrinone), and the new biologic agents infliximab and rituximab. Heparin causes an immune thrombocytopenia that leads to severe thrombosis rather than bleeding, through a mechanism different from other DITPs.
Incidence
DITP incidence is not well defined and is reported to be low, ranging from 0.6 to 1.6 per 100,000. These studies used national surveillance programs and hospital records, and probably underestimate the frequency of DITP. However, the incidence of thrombocytopenia induced by GPIIb/IIIa inhibitors (tirofiban, eptifibatide, and abciximab) and heparin is higher. DITP-associated GPIIb/IIIa inhibitors have an incidence of 0.1% to 2% on first drug exposure and about 5% to 10% on subsequent exposures. This incidence suggests that thousands of patients globally must develop this drug complication every year because millions of cardiac patients receive these drugs for prevention of coronary artery thrombosis following coronary angioplasty, stent insertion, and acute coronary syndrome. Despite the variable frequencies, DITP represents a significant clinical problem because of the large number of drugs involved and the large number of patients who can be affected.
Pathophysiology
DITP is usually mediated by a drug-dependent antibody that binds to platelets or megakaryocytes (Mks) only in the presence of the sensitizing drug. Over the years, several hypotheses have been proposed for the antibody-platelet interactions. These hypotheses are discussed later ( Tables 1 and 2 ).
| Major Mechanisms | Hypotheses |
|---|---|
| Increase in peripheral platelet destruction or clearance | Increased platelet clearance by splenic and hepatic macrophages Platelet lysis by complements in circulation Platelet lysis by free oxygen radicals in circulation |
| Suppression of platelet production | Suppression by megakaryocyte proliferation and differentiation Increase in megakaryocyte apoptosis Inhibition of proplatelet production Suppression of megakaryocyte GPIb-IX expression |
| Hypotheses | Descriptions |
|---|---|
| Quinine-type DITP | Drug binds to a hyperflexible domain of a platelet glycoprotein (GPIX or GPIIIa) and the antibody recognizes an epitope on the GP. There are 2 proposed models:
|
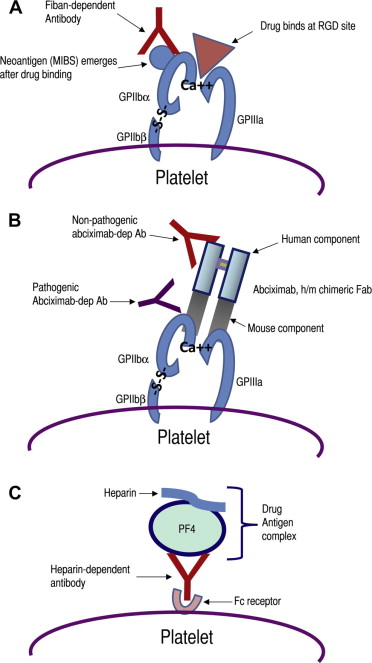
| Fiban-induced DITP | Fiban drug binds to GPIIb or GPIIIa near the RGD binding site, inducing a conformational change and formation of a neo-epitope (MIBS; see text) to which the antibody binds (see Fig. 3 A) |
| Abciximab-induced DITP | The patient’s antibodies recognize and bind to a mouse sequence of abciximab chimeric Fab (see Fig. 3 B) |
| Immune-complex mechanism | The antibody binds to a plasma protein-drug complex (with drug acting as a hapten) and immune complex binds to platelet via its Fc receptor and the platelet is destroyed/activated as an innocent bystander (see Fig. 3 C) |
Hapten-induced antibody or hapten mechanism
Early immunologic studies in the 1900s suggested that small molecules like drugs are not antigenic and can only trigger an immune response when linked covalently to a macromolecule (a protein) acting as a hapten. The resulting antibodies recognize the carrier molecule only where the hapten is attached covalently. In the 1950s and 1960s, Shulman proposed the innocent-bystander or immune-complex mechanism that replaced the hapten mechanism as the dogma, and it remained accepted until the 1980s. According to the immune-complex mechanism, drug-antibody complexes bind to platelets via platelet Fc receptors. Platelets are targeted for destruction as innocent bystanders as the antibody recognizes the drug but not the platelet. It is now well recognized that the hapten and the immune-complex mechanisms are relevant for only a few drugs: high-dose penicillins cause immune hemolysis by haptens and heparin causes thrombocytopenia via immune-complex binding. These mechanisms are not discussed further in this article. Heparin-induced thrombocytopenia (HIT) represents a unique clinical-pathologic entity with strong platelet activation and frequent thrombosis. HIT merits a separate discussion with a full article to itself.
Quinine-type immune thrombocytopenia
As stated earlier, thrombocytopenia caused by quinine or its isomer, quinidine, is the prototype of most DITPs. This mechanism or a variation thereof mediates immune thrombocytopenia caused by most drugs (see Table 2 ). In 1983, we showed for the first time that, using staphylococcal protein A, we could immunoprecipitate the drug-platelet antigen (glycoprotein Ib-IX [GPIb-IX]) complex with the antibody of a patient with quinidine-induced thrombocytopenia. This radical finding contradicted the immune-complex mechanism dogma at that time, because this finding suggests that the antibody binds to platelets by its Fab but not its Fc domain. This finding was confirmed by subsequent studies from our and other laboratories. It became clear that the immune-complex mechanism does not contribute to thrombocytopenia caused by quinine, quinidine, or most other drugs, except heparin.
Current evidence suggests that the drug binds to platelet GPIb-IX (von Willebrand factor receptor complex) and/or GPIIb/IIIa (fibrinogen receptor complex).
One hypothesis (model 1, Fig. 1 A ) is that drug binding induces a conformational change of the platelet surface protein, resulting in the emergence of a cryptic epitope or neoantigen previously unseen by the immune system. The resultant antibody recognizes and binds this epitope only in the presence of the drug. In the absence of the drug, this epitope or neoantigen is buried in the glycoprotein folds or does not exist. Aster and colleagues recently proposed a new model (model 2, see Fig. 1 B) that is slightly different from the model discussed earlier. The essential difference is that, unlike model 1 in which the drug and the antibody bind the glycoprotein at different sites, in model 2 the drug and the antibody bind the target platelet glycoprotein at the same site (the drug is considered an essential component of the antibody binding site). Model 2 is supported by observations that quinine bound to platelets is more resistant to removal in the presence of the drug-dependent antibody, suggesting that the drug may be part of the binding pocket and is, therefore, entrapped by the bound antibody. Nevertheless, there is no conclusive or direct experimental evidence to support this model.
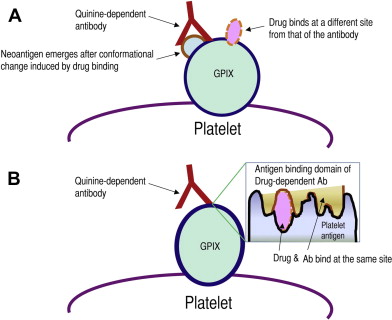
Why do the drug-dependent antibodies target specific platelet glycoproteins such as GPIb-IX?
We have consistently found that all patients with quinine-induced and quinidine-induced thrombocytopenia had antibodies that reacted drug dependently with GPIb-IX ; almost all of these antibodies recognized GPIX and only an occasional antibody had specificity for GPIbα. In addition, a minority of patients (<30%) had drug-dependent antibodies reactive with GPIIb-IIIa. In contrast, other investigators found a higher proportion of anti–GPIIb-IIIa antibodies. Furthermore, we have mapped the binding site of the GPIX-specific quinine-dependent antibodies to a restricted site in the membrane-proximal ectodomain of platelet GPIX (R110–Q115) and the GPIbα-specific antibody to the N-terminal domain of GPIbα (residues 283–293).
Peterson and colleagues located the binding sites of the quinine-dependent antibodies to a 29-kDa region of GPIIIa comprising the hybrid and the plextrin-semaphorinintegrin (PSI) domains, and residues A50 and D66 played a critical role in the formation of the antibody binding sites. Patients with rifampicin-induced and ranitidine-induced thrombocytopenia also have antibodies that react with GPIX. An anti-GPIX monoclonal antibody (mAb) SZ1 cross-blocked the binding of quinine-dependent, quinidine-dependent, rifampicin-dependent, and ranitidine-dependent antibodies to GPIX, suggesting that these four antibodies bind to a site that coincides with or is close to the epitope of SZ1. It is possible that, when other less well-characterized drug-induced antibodies (eg, vancomycin-induced antibody) are studied, they may also bind to this restricted GPIX site. These data also suggest that this region must have unique characteristics that render it unusually antigenic on drug binding.
Flexible domains and immunogenicity
Protein structure studies have recently provided additional information to refine the current hypothesis. The crystal structure of GPIIIa showed that the PSI domain (which is targeted by quinine-dependent antibodies) could not be completely resolved, indicating that it possesses a degree of mobility (hyperflexibility), adopting several transient state conformations. The membrane-proximal part of the ectodomain of GPIX is also highly flexible and is likely to adopt a range of non-native or denatured states (model in Fig. 2 ). We have mapped the binding sites of anti-GPIX quinine-dependent antibodies to this hyperflexible GPIX region as described earlier.
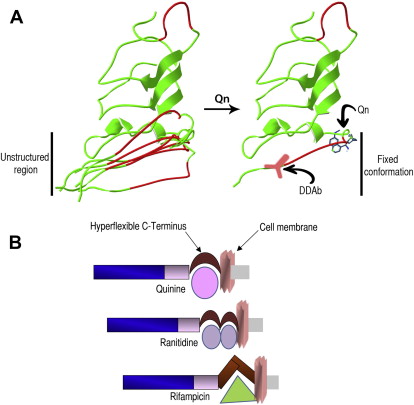
The presence of these unstructured protein domains may allow the binding of drugs of diverse chemical structures as the protein changes from one non-native conformation to another (see Fig. 2 ). Non-native conformations are characterized by an increase in their hydrophobic surface, which may facilitate interaction by drugs containing hydrophobic pockets, such as quinine and quinidine, and it could also explain why drugs of different chemical structure, such as quinine, rifampicin, and ranitidine, interact with GPIX or GPIIIa. It is likely that each drug binds to distinct non-native structures, which then adopt a particular conformation. Under some conditions, this fixed configuration may elicit an immune response resulting in the generation of drug-dependent antibodies (see Fig. 2 ).
Thrombocytopenia induced by GPIIb/IIIa inhibitors (fiban)
GPIIb/IIIa inhibitors, tirofiban and eptifibatide (fiban), are synthetic small molecules that bind tightly to the arginine-glycine-aspartic acid (recognition sequence arginine-glycine-aspartic acid [RGD]) recognition site on GPIIb/IIIa, inhibit fibrinogen binding, block platelet aggregation, and prevent platelet thrombus formation. These drugs are widely used in clinical medicine to reduce cardiac complications following percutaneous transluminal coronary angioplasty (PTCA) and to treat acute coronary syndrome. Clinical trials and subsequent clinical experience with the fibans showed that about 0.2% and 2% of patients receiving these compounds developed an acute, frequently severe, thrombocytopenia (platelet <10 × 10 9 /L) within a few hours of first drug exposure. Previous studies have shown that naturally occurring antibodies against GPIIb/IIIa cause the thrombocytopenia, thus explaining the occurrence of thrombocytopenia on first drug exposure. These antibodies are found not only in patients with fiban-induced thrombocytopenia (FIT) but also in normal volunteers never exposed to the drug. It is still uncertain whether healthy individuals with these naturally occurring antibodies will develop severe thrombocytopenia if they receive a fiban. If so, detection of the antibody before drug administration may prevent this serious complication.
Integrin research has revealed that RGD peptide and RGD mimetic compounds induce conformational changes in GPIIb/IIIa, resulting in the emergence of cryptic integrin domains that could be recognized by monoclonal antibodies specific for these ligand-induced binding sites (LIBSs). It has been suggested that antibodies from patients with FIT probably have specificity for these LIBSs. Until recently, it was uncertain whether the ligand-induced GPIIb/IIIa determinants that resulted from fibrinogen/RGD peptide binding (ie, LIBSs) are the same as those induced by fiban drugs. Bougie and colleagues showed that LIBSs are different from mimetic-induced binding sites (MIBSs), a term they used for determinants induced by fiban, but not fibrinogen, binding to GPIIb/IIIa. They also found that FIT antibodies did not recognize LIBS determinants as previously expected but instead recognized conformational changes in GPIIb/IIIa induced by the fiban drugs (MIBSs). In most patients with eptifibatide-induced and tirofiban-induced thrombocytopenia, antibody binding was drug specific and occurred only when the integrin complexed with the sensitizing drug. However, antibody cross-reactivity occurred only in a minority of patients. These investigators also reported that the MIBSs resided in the head region of GPIIb/IIIa, near the RGD recognition site, probably on the β-propeller domain of GPIIb or the βA domain of GPIIIa ( Fig. 3 A ). More precise epitope mapping, particularly identification of GPIIb or GPIIIa sequences that mediate antibody binding, may have to await further studies.
Abciximab-induced thrombocytopenia
Abciximab is a chimeric (mouse/human) monoclonal antibody (Fab fragment) that has specificity for the βA domain of GPIIIa. Because abciximab binds close to the RGD recognition site of GPIIIa, it blocks fibrinogen binding to activated GPIIb/IIIa and hence inhibits platelet thrombus formation. About 1% to 2% of patients given the drug develop acute, often severe, thrombocytopenia on first drug exposure, and 10% to 12% on the second and subsequent exposure. Like FIT, naturally occurring abciximab-dependent antibodies were detected in patients receiving the drug and in healthy volunteers never previously exposed to abciximab. There is a subgroup of patients who developed acute thrombocytopenia about a week after infusion of the drug. These patients presumably did not have naturally occurring antibodies and acquired the antibody on drug sensitization. The antibody reached a level that could react with the abciximab that remained on platelets approximately 7 days later.
Unlike other DITPs, both abciximab-dependent antibodies recognize the drug (abciximab) when bound to platelets. However, the patient antibody recognizes a mouse domain in the chimeric Fab. In contrast, the antibody in healthy individuals is specific for the papain cleavage site introduced during the manufacturing process and, consequently, binding can be inhibited by Fab fragments prepared from normal immunoglobulin (Ig) G (see Fig. 3 B).
How may drug-dependent antibodies cause thrombocytopenia?
Platelet Destruction or Clearance
Thrombocytopenia occurs when there is an increase in platelet clearance/destruction or a decrease in platelet production in the bone marrow. The most widely held view is that in DITP, like other immune thrombocytopenias, antibody-coated platelets are cleared in the spleen and/or liver by macrophage phagocytosis. However, there is little or no direct evidence for this. We and Bougie and colleagues recently provided in vivo evidence using a nonobese diabetic/severe combined immunodeficiency (NOD/SCID) mouse model. We were able to show that human platelets injected immediately after administration of DITP serum or purified IgG resulted in rapid platelet clearance. This rapid and marked platelet clearance was uniformly seen across 7 patients with quinine-induced thrombocytopenia, consistent with the clinical picture of severe thrombocytopenia of abrupt onset in this condition. In comparison, platelet clearance using this in vivo model by antibodies from patients with primary immune thrombocytopenia (ITP) is gradual and variable in rate and extent, in keeping with the heterogenous clinical picture in ITP. Additional mechanisms have been proposed for the thrombocytopenia in DITP; these include platelet destruction in circulation by complement-mediated or radical oxygen species (ROS)–mediated lysis, but there is no direct evidence for these hypotheses.
Early studies in the 1950s showed in vitro complement deposition on platelets following DITP antibody-platelet interaction. Platelet lysis caused by oxidative stress has been shown in immune thrombocytopenia associated with human immunodeficiency virus (HIV) infection. Platelet lysis by cytotoxic T cells has also been implicated in platelet number decline in ITP. There is no convincing or direct evidence of complement-mediated, ROS-mediated, or T-cell–mediated platelet destruction in DITP.
Suppression of Platelet Production
Despite evidence that antibodies of patients with DITP can directly destroy platelets in vivo, there is considerable support for additional platelet-independent processes that may contribute to the thrombocytopenia in DITP. One of these is antibody-mediated megakaryocytic damage. Most experimental data in this aspect come from studies in patients with ITP or using antiplatelet monoclonal antibodies. However, there is also recent evidence indicating that this is also applicable to DITP.
In the 1970s, McMillan and colleagues observed that antiplatelet antibodies produced by patients with ITP could recognize Mks. Subsequent experiments suggested a possible role of Mks in the pathogenesis of ITP. These ideas were strengthened by Chang and colleagues who showed that anti-GPIb/IX ITP antibodies affected Mk differentiation, and by McMillan and colleagues who showed a reduction in Mk production after in vitro culture of Mks in the presence of antiplatelet antibodies. These investigations implied that megakaryocytic damage by antiplatelet antibodies may result in a decrease in platelet production and consequently thrombocytopenia.
DITP antibodies causing Mk damage have been shown in vitro using serum from a patient with eptifibatide-induced thrombocytopenia. Furthermore, inhibition of Mk differentiation and proliferation together with decreased platelet production capacity were recently shown in a more in-depth study using IgG/sera from patients with quinine-induced thrombocytopenia antibodies. Therefore, DITP antibodies may also contribute to thrombocytopenia by suppressing platelet production.
Related posts:
 Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
 Platelet Transfusion Therapy
Diagnosis and Management of Heparin-Induced Thrombocytopenia
Platelet Transfusion Therapy
Diagnosis and Management of Heparin-Induced Thrombocytopenia
 Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
Diagnosis and Management of Heparin-Induced Thrombocytopenia
Harnessing the Platelet Signaling Network to Produce an Optimal Hemostatic Response
Diagnosis and Management of Heparin-Induced Thrombocytopenia
 Thrombotic Thrombocytopenic Purpura and the Atypical Hemolytic Uremic Syndrome
Thrombotic Thrombocytopenic Purpura and the Atypical Hemolytic Uremic Syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


