Introduction
Alzheimer’s disease (AD) mainly affects elderly individuals. Because of the ageing of populations worldwide, this disorder is reaching epidemic proportions, with a large human, social and economic burden. The progressive degeneration of neurons in AD causes abnormalities of the systems of neurotransmitters, and a combination of cholinergic and glutamatergic dysfunction appears to underlie the symptomatology of AD. Current drugs for AD target cholinergic and glutamatergic neurotransmission. Cholinesterase inhibitors (ChEIs) and/or butyrylcholinesterase are widely prescribed as symptomatic treatments for AD. Donepezil (Aricept), rivastigmine (Exelon) and galantamine (Reminyl) were approved by the US Food and Drug Administration (FDA) for the treatment of mild to moderately severe AD in 1996, 2000 and 2001, respectively. The use of donepezil for the treatment of severe AD has already been approved in the USA. Memantine [Ebixa and Axura (Europe) and Namenda (USA)] was approved in 2002 by the European Agency for the Evaluation of Medical Products (EMEA) for the treatment of moderately severe to severe AD and in 2003 by the FDA for the treatment of moderate to severe AD.
However, the effects of these treatments are limited or controversial as they do not modify disease progression.1 Currently, much effort is directed towards identifying disease-modifying therapies. The use of biomarkers of plasma, cerebrospinal fluid (CSF) or neuroimaging [magnetic resonance imaging (MRI) and positron emission tomography (PET)] could play an important role in estimating their efficiency and their potential disease-modifying effect.
This chapter discusses general classes of potential disease-modifying drugs under investigation for the treatment of AD, and also the contribution of the non-pharmacological approach. Compounds and studies selected for this review were identified by systematic searches using PubMed. Only publications in English were reviewed. The ClinicalTrials.gov (http://www.clinicaltrials.gov) website was used for information on ongoing randomized controlled trials (RCTs).
Impact on Cholinergic Deficit
The mainstays of current pharmacotherapy for AD are compounds aimed at increasing the levels of acetylcholine (ACh) in the brain, thereby facilitating cholinergic neurotransmission through inhibition of the cholinesterases. Other drugs that can increase the ACh levels in brain include ACh precursors, muscarinic agonists and nicotinic agonists.
Recent reports suggest that AChEIs could affect the underlying disease processes2, 3 through neuroprotective and disease-modifying properties.4 Huperzine A is a selective AChEI with potential properties that include modification of β-amyloid (Aβ) peptide processing, reduction of oxidative stress, neuroprotection and regulation of nerve growth factor (NGF) expression.5, 6 Clinical trials of its derivative, ZT-1, have demonstrated an improvement in cognitive function of AD patients.7 Phenserine, a derivative of physostigmine, has a dual mode of action: AChEI and inhibitor of the formation of Aβ precursor protein (APP).8, 9 Phenserine is dose-limited in animals by its cholinergic actions. The (+)-phenserine enantiomer (Posiphen), which has weak activity as an AChEI and is potent on Aβ levels and amyloid processing, can be dosed much higher,10, 11 but clinical trials are required. Butyrylcholinesterase may play a role in attention, executive function, emotional memory and behaviour. Furthermore, butyrylcholinesterase activity progressively increases as the severity of dementia advances, whereas acetylcholinesterase activity declines. Therefore, inhibition of butyrylcholinesterase may provide additional benefits.12 Structural analogues of phenserine, cymserine and bisnorcymserine, proved to be potent inhibitors of human butyrylcholinesterase in comparison with phenserine.13, 14 Salvia officinalis (sage), which has cholinergic properties, is also under clinical investigation for AD.
Several M1 receptor agonists, such as Lu 25–109 (a compound that directly stimulates muscarinic cholinergic receptors), have been tested in clinical trials without much success.15 Recent studies which suggest the role of muscarinic agonists in regulating the production of Aβ again raise the possibility that selective M1 agonists could be useful in AD.16 The M1 muscarinic agonists are neurotrophic, elevate the nonamyloidogenic APP in vitro, decrease Aβ levels in vitro and in vivo and restore cognitive impairments in animal AD models.17, 18
Nicotinic acetylcholine receptors (nAChRs), which are essential for learning and memory, are reduced in AD brains and research implicates a role for nAChRs in neuroprotection. Targeting nAChRs is an attractive therapeutic approach and several selective ligands for nAChRs have been developed, but a challenge has been the reduction of side effects.19–22 ABT-089, a selective neuronal nicotinic receptor modulator which shows positive effects in rodent and primate cognitive models,23 is a candidate for further evaluation as a treatment for AD.24 GTS-21 (DMXBA) is a selective agonist of alpha7 nicotinic receptors which enhances a variety of cognitive behaviours in mice, monkeys, rats and rabbits. It also displays neuroprotective activity in vitro and has shown promising characteristics during phase 1 clinical tests.25 Ispronicline (TC-1734, AZD-3480) is a selective neuronal nicotinic agonist that is neuroprotective in vitro and exhibits memory-enhancing properties in vivo. Ispronicline also had a beneficial effect on cognition in subjects with age-associated memory impairment in a phase 2 trial.26 Other nicotinic agents under clinical evaluation are summarized in Table 78.1.
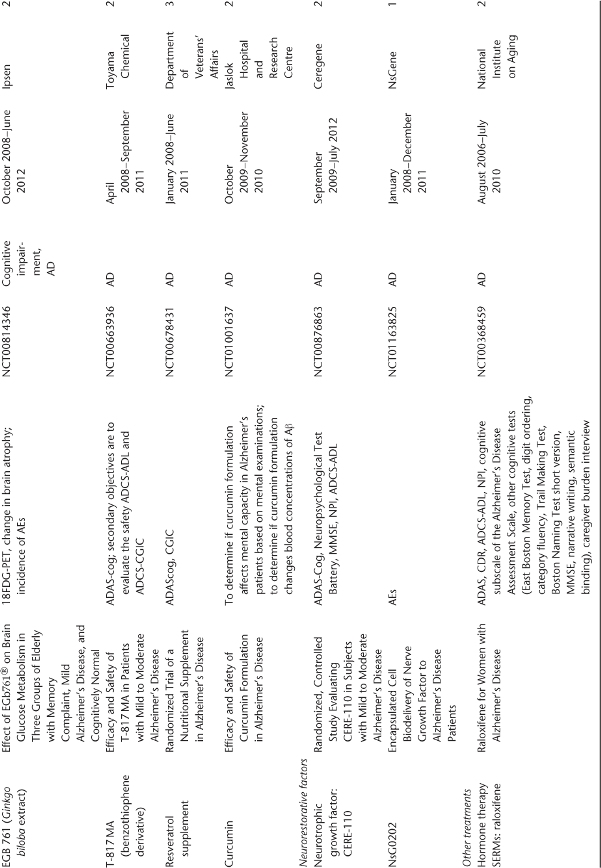
Table 78.1 Ongoing clinical research on new drugs (based on ClinicalTrials.gov website)a.
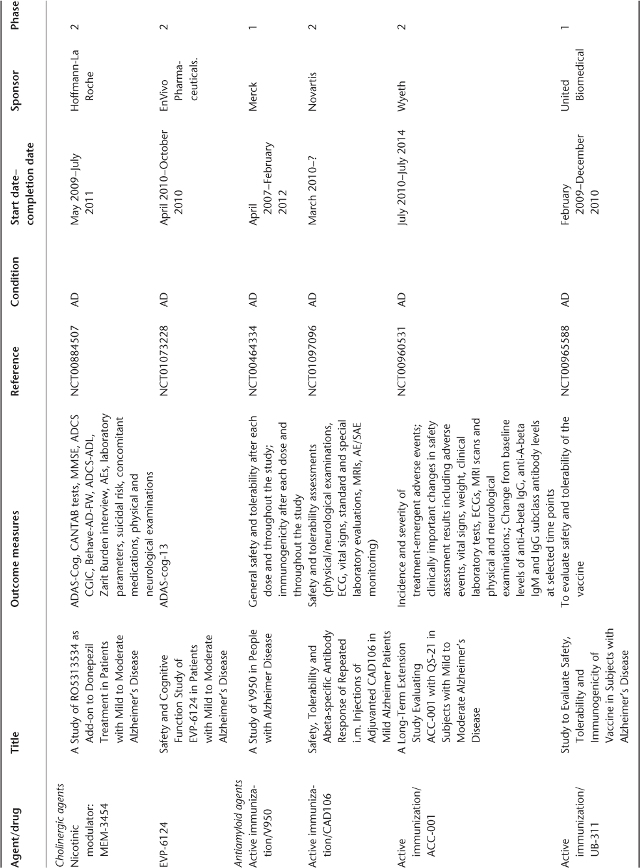
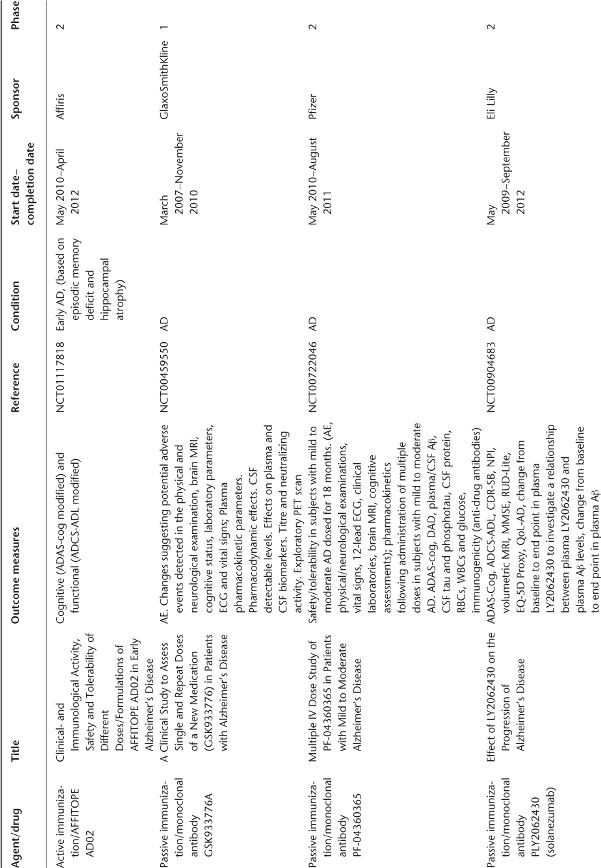
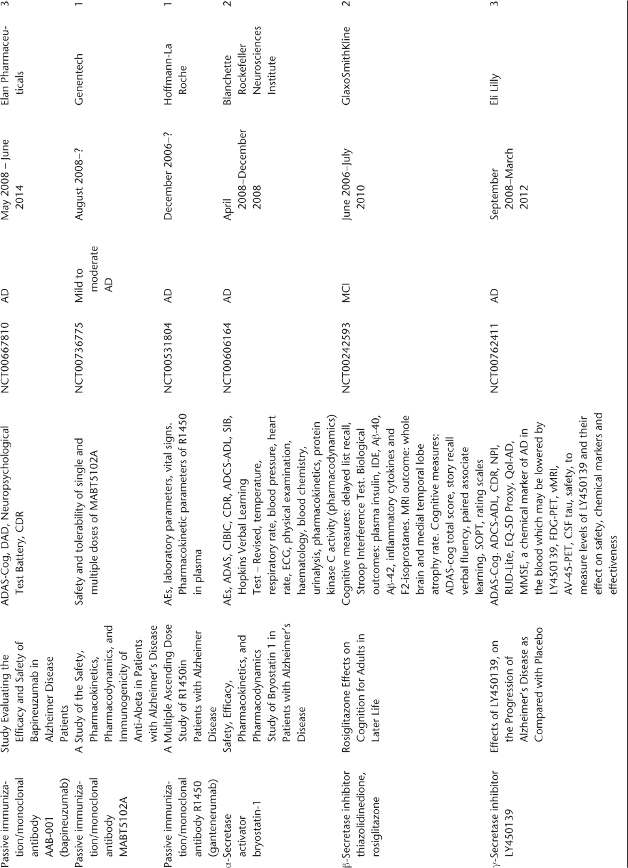
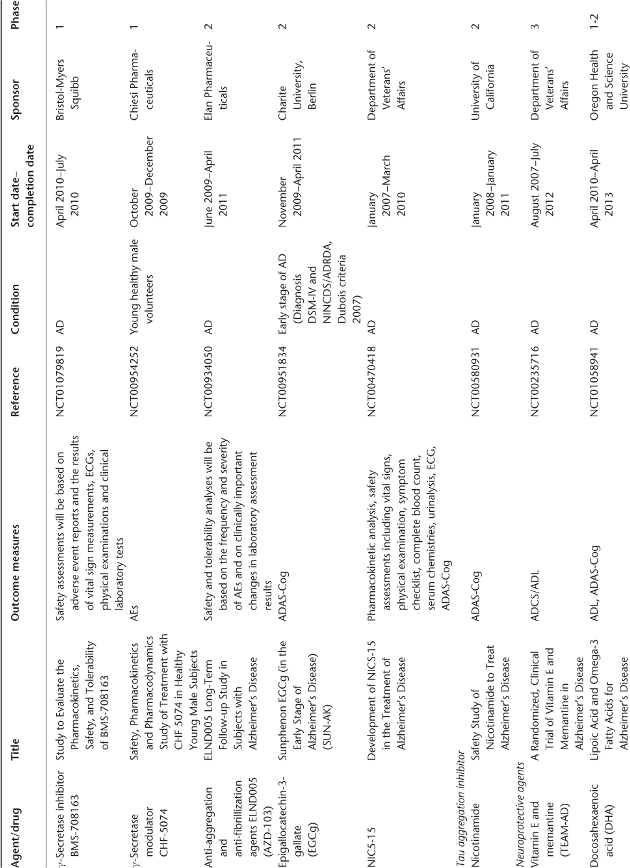

Anti-Amyloid Therapies
AD drug development is driven mainly by the amyloid hypothesis. In fact, currently available evidence strongly supports the position that the initiating event AD is related to abnormal processing of β-amyloid (Aβ) peptide, ultimately leading to the formation of Aβ plaques in the brain.27 This process occurs while individuals are still cognitively normal. After a lag period, which varies from patient to patient, neuronal dysfunction and neurodegeneration become the dominant pathological processes. Anti-amyloid agents target production, accumulation, clearance or toxicity associated with Aβ peptide.
Drugs to Promote Aβ Clearance
Active and passive immunizations were developed to inhibit generation of toxic Aβ aggregates and to remove soluble and aggregated Aβ.
Active Immunization
Active immunization of APP transgenic (Tg) mice before they had amyloid plaque deposits resulted in significantly reduced amyloid deposits and neuritic pathology, and Aβ immunization of older mice with pre-existing plaques also resulted in a reduction in plaque pathology. This suggests that this approach is able to slow the progression of amyloid deposition and even reverse it.28 Subsequent studies have shown that Aβ immunization can also prevent or improve learning deficits in AD Tg mice.29, 30 A phase 1 human trial using an active immunization strategy against Aβ was promising but the phase 2a immunization trial with a synthetic Aβ peptide called AN-1792 was stopped after reports of meningoencephalitis in 6% of the treated patients.31 The first analysis of efficacy in this trial, reported for a small subset of patients, was suggestive of a slowing cognitive decline, particularly in patients generating the highest antibody titres.32 A more recent and complete analysis of all treated patients demonstrated no significant efficacy except in the small subset of subjects who had CSF examinations, CSF tau was decreased in antibody responders versus placebo subjects (p < 0.001).33 Immunization strategies research in Tg mouse models has been refocused to establish safer therapeutic approaches.34 A new generation of AD vaccines has been designed to try to prevent the induction of Aβ reactive T cells, which is thought to have been critical in AN-1792 failure.35
Passive Immunization
It is also possible to bypass the immune response by direct administration of anti-Aβ antibodies with a passive immunization approach. This approach seems to be as effective as active immunization in Tg mice36 and could potentially eliminate toxic T-cell-mediated responses to Aβ. In preclinical studies, passive immunization of APP Tg mice with pre-existing evidence of cerebral amyloid angiopathy resulted in an increased severity and incidence of microhaemorrhages, but the physiological implications of these findings remain unclear.37, 38 Antibodies against the β-secretase cleavage site of the APP are another way to limit APP processing. They inhibit Aβ formation in vitro and their long-term administration to Tg mice improved cognitive functions associated with a reduction in brain inflammation and incidence of microhaemorrhage.39 Classical human immunoglobulin (Ig) preparation can also be investigated as a passive immunization therapy in AD, as a small percentage of antibodies are directed against Aβ peptide sequences. Intravenous infusion of Igs in five AD patients over a 6 month period prevented further cognitive decline,40 suggesting that this approach could potentially act like a passive immunotherapy. Human trials of passively administered anti-Aβ antibodies are now being initiated. Other antibodies have recently reached clinical evaluation: GSK933776A, PF-04360365, PLY2062430 (solanezumab) and AAB-001 (bapineuzumab) (Table 78.1). In a recent RCT that enrolled patients with mild-to-moderate AD, treatment with bapineuzumab for 78 weeks reduced cortical 11C-PiB retention compared with both baseline and placebo.41
Drugs to Reduce Aβ Production
α-Secretase Activators
APP processing by α-secretase is a non-amyloidogenic pathway, because the α-secretase cleavage site is within the Aβ sequence of APP. Enhanced cleavage at this site may represent a potential disease-modifying strategy. Bryostatin 1, a macrolide lactone, exhibits high affinity for protein kinase C and dramatically enhances the secretion of the α-secretase product in patients’ fibroblasts42 (Table 78.1). Etazolate (EHT-0202), a selective GABA receptor modulator, stimulates neuronal α-secretase and increases sAPPα production.43 This drug, which is orally bioavailable, has recently been tested in a phase 2 RCT in patients with mild-to-moderate AD (NCT00880412, results not available). Talsaclidine, an M1 agonist that stimulates the non-amyloidogenic α-secretase processing in vitro and decreases CSF Aβ in AD patients following chronic treatment,44, 45 holds potential disease-modifying properties.
β-Secretase Inhibitors
β-secretase has been shown to be a transmembrane aspartic protease, β-site Aβ cleaving enzyme 1 (BACE1). BACE-1 processing of Aβ precursor protein is the first step in the pathway leading to the production of Aβ. BACE-1 knockout mice develop normally and appear to have completely abolished Aβ production.46 A selective BACE-1 inhibitor, GSK188909, reduced levels of secreted and intracellular Aβ40 and Aβ42 in vitro and also in APP transgenic mice brains.47 Thiazolidinediones also act as β-secretase inhibitors by stimulating peroxisome proliferator-activated receptor-γ (PPARγ). PPARγ agonists, such as rosiglitazone, also have anti-inflammatory effects.48, 49 Thiazolidinediones are under evaluation in AD, but recent data have shown a potential higher risk of myocardial infarction with these compounds.50 To compensate for the brain’s reduced ability to use glucose in AD, administration of ketone bodies or their metabolic precursors such as medium-chain triglycerides (MCTs) might be another strategy. In a preliminary study with 20 subjects with AD or mild cognitive impairment (MCI), single doses of MCTs demonstrated pharmacological activity and significant efficacy in cognitive performance.51 A phase 2b clinical trial in AD patients confirmed ketone bodies’ safety and efficacy on cognition. A pivotal phase 3 clinical trial in AD patients is planned.52
γ-Secretase Inhibitors and Modulators
Inhibition of γ-secretase targets the generation of Aβ42, but other proteins are also substrates of this enzyme and particularly the transmembrane Notch receptor, involved in vital functions.53–55 Abnormalities in the gastrointestinal tract, thymus and spleen in animal models result from inhibition of Notch cleavage.56, 57 Preclinical studies established that γ-secretase inhibitors can reduce brain Aβ and reverse Aβ-induced cognitive deficits in transgenic mice. LY450139 dihydrate, a γ-secretase inhibitor, inhibits Aβ formation in vitro and in vivo. In phase 1 volunteer studies, a dose-dependent reduction in plasma Aβ was demonstrated. However, Aβ concentrations were unchanged in CSF.58, 59 In an RCT of AD patients treated with LY450139 dihydrate, Aβ40 decreased significantly in plasma and decreased in a non-significant manner in CSF.60 Single doses of GSI-953, a selective γ-secretase inhibitor, also produced dose-dependent reductions of plasma but not CSF Aβ peptides in humans.61 Tarenflurbil is the pure R-enantiomer of flurbiprofen and is the first in a novel class of selective Aβ-42-lowering agents. It modulates γ-secretase and is highly specific for its effects on Aβ-42 and, unlike the γ-secretase inhibitors, does not interfere with the function of Notch. In a phase 2 study, tarenflurbil was well tolerated for up to 24 months of treatment in 210 AD patients, with evidence of a dose-related effect on measures of daily activities and global function in patients with mild AD,62 but a phase 3 clinical trial was negative. Table 78.1 gives an overview of drugs that are currently in research and development in this field.
Anti-Aggregation and Anti-Fibrillization Agents
An alternative approach to secretase inhibition, which raises the problem of interfering with normal enzymatic reactions, is to inhibit Aβ aggregation into neurotoxic oligomers. Tramiprosate (NC-531 or 3APS) is a glycosaminoglycan mimetic that binds to Aβ and inhibits amyloid plaque formation.63 Preclinical data have shown that tramiprosate reduces brain and plasma levels of Aβ and prevents fibril formation.64 In a phase 2 trial, long-term administration of tramiprosate was safe, well tolerated and reduced CSF Aβ-42 levels in patients with AD. Tramiprosate has reached phase 3 clinical trials.65 However, a phase 3 trial in the USA was negative and was stopped in Europe. Dysregulation of cerebral metal ions (Fe2+, Cu2+ and Zn2+) and their interactions with Aβ may contribute to AD by playing a role in the precipitation and cytotoxicity of Aβ.66 Metal ions are required for Aβ protein oligomerization and recent studies showed that metal chelators could produce a significant reversal of Aβ deposition in vitro and in vivo.67 XH1 and DP-109, both metal chelators, attenuated Aβ pathology in APP Tg mice.68, 69 Clioquinol (PBT-1) is a Cu/Zn chelator that promotes Aβ dissolution. In a pilot phase 2 clinical trial in AD patients, this fibrillization inhibitor shows a significant efficacy in the more severely affected group according to the authors70 but this point is discussed.71 Another chelator, desferioxamine, has shown some benefit in AD, but also severe adverse effects.72 PBT-2, another metal protein-attenuating compound, was tested in a phase 2 trial in patients with early AD. PBT-2 affects the Cu2+-mediated and Zn-mediated oligomerization of Aβ protein. The safety profile was favourable. Cognitive efficacy was restricted to two measures of executive functioning. The effect on putative biomarkers for AD in CSF but not in plasma was suggestive of a central effect of the drug on Aβ metabolism.73 Another compound interfering with the aggregation and fibrillization of Aβ, ELND005 (AZD-103), is under evaluation in a clinical trial (Table 78.1).
Drugs to Target Tau Protein
Microtubule-associated protein (MAP) tau is abnormally hyperphosphorylated in AD. Several kinases are reported to phosphorylate tau in vitro, including glycogen synthase kinase-3 (GSK-3), cyclin-dependent kinase-5 (CDK-5), mitogen-activated protein kinase family members (MAPK), casein kinase, calcium calmodulin-dependent kinase II, protein kinase A and others. Some of them, such as GSK-3, could be also involved in Aβ generation, promoting cell death, production of inflammatory molecules and cell migration.74 Phosphoseryl/phosphothreonyl protein phosphatase-2A (PP-2A), which is co-localized with tau and microtubules in the brain, is apparently the most active enzyme in dephosphorylating the abnormal tau to a normal-like state.75 Other phosphatases have also been implicated.76 Reducing abnormal phosphorylation and restoring or stimulating phosphatase activity are promising therapeutic strategies. Lithium reduces tau phosphorylation in vitro, promotes microtubule assembly through inhibition of GSK-377 and has been shown to reduce tau phosphorylation in APP Tg mice.78 In a preliminary clinical trial of lithium in AD patients, no effect of lithium on tau and Aβ-42 in the CSF was observed, which does not support the notion that lithium may lead to reduced hyperphosphorylated tau in AD after short-term treatment.79 Methylthioninium chloride (Trx0014) has been shown in vitro to prevent aggregation of tau into tangles. It has demonstrated cognitive and behavioural benefits in animal models.80
Neuroprotective Agents
Another alternative approach involves protection against cellular damage caused by oxidative, inflammatory or other toxic stressors.
Antioxidants
Genetic and lifestyle-related risk factors for AD could be associated with an increase in oxidative stress, suggesting that oxidative stress is involved in the early stage of the pathology.81 Individuals with MCI or very mild AD show increased levels of lipid peroxidation and nucleic acid oxidation in postmortem brain and plasma.82 Free radicals and oxidative injury to neurons could chronologically precede Aβ plaque deposition and tau phosphorylation.83 Several antioxidants that have been investigated for their potential to reduce the risk of AD include vitamins A, C and E, coenzyme Q, selenium and polyunsaturated fatty acids. Vitamin E had been show to slow progression of the disease in patients with moderately severe AD.84 However, recent meta-analysis and trial results suggest that vitamin E increases morbidity and mortality85 and a Cochrane review does not support the use of vitamin E to treat AD.86 The lack of consistent efficacy data for vitamin C and its questionable safety could also discourage its use.87 Most of the published epidemiological studies are consistent with a positive association between high reported omega-3 polyunsaturated fatty acid consumption and a lower risk of developing cognitive decline or AD later in life.88 Docosahexaenoic acid (DHA) is the most abundant omega-3 fatty acid in the brain. DHA acts in the brain via neurotrophic and anti-apoptotic pathways. In addition, DHA may act through anti-neuroinflammatory pathways, as DHA possesses anti-inflammatory properties in the periphery. The results from the first randomized, double-blind, placebo-controlled clinical trial evaluating the effects of dietary omega-3 fatty acid supplementation on cognitive functions in patients with mild to moderate AD showed no significant efficacy of daily intake of DHA and eicosapentaenoic acid. However, in a subgroup of patients with very mild cognitive dysfunction [Mini Mental State Examination (MMSE) >27 points], a significant reduction in MMSE decline rate was observed in the omega-3 fatty acid-treated group compared with the placebo group.89 α-Lipoic acid (LA), an essential cofactor in mitochondrial dehydrogenase reactions, functions as an antioxidant and reduces oxidative stress.90 LA seems to exert a cellular protective effect as evidenced by decreases in apoptotic markers in fibroblasts from AD patients.91 Clinical preliminary data show that LA might be a successful therapy for AD.92 EGb761, a Ginkgo biloba extract that has free radical scavenging properties, inhibits the formation of Aβ fibrils, attenuates mitochondrion-initiated apoptosis and decreases the activity of caspase-3, a key enzyme in the apoptosis cell signalling cascade.93 Mitoquinol, an antioxidant that targets mitochondrial dysfunction, has demonstrated encouraging preclinical results. Mitoquinol mimics the role of the endogenous mitochondrial antioxidant coenzyme Q10 and augments its antioxidant capacity to supraphysiological levels.94 Melatonin, an indolamine secreted by the pineal gland, may also protect neuronal cells from Aβ-mediated toxicity via antioxidant properties and could attenuate tau hyperphosphorylation.95 Isoflavones are also under clinical evaluation for their antioxidant properties (Table 78.1). Other clinical trials with antioxidants are on the way (Table 78.1).
Anti-Inflammatory Drugs
Laboratory evidence shows that inflammatory mechanisms contribute to neuronal damage in AD. Epidemiological evidence96 suggests that non-steroidal anti-inflammatory drugs (NSAIDs) may favourably influence the course of the disease. In a 1993 trial, indomethacin appeared to protect AD patients from cognitive decline, according to the authors,97 but this point of view is not shared by Cochrane reviewers.98 Another trial with indomethacin failed to show any efficacy in the progression of AD.99 Ibuprofen, celexocib, rofecoxib and naproxen did not slow the progression of AD.100–102 In a phase 2 AD clinical trial with (R)-flurbiprofen, a few subsets of patients who had high blood concentrations of this drug demonstrated a benefit in cognitive and behavioural performance.103 However, Myriad Genetics has discontinued the development of (R)-flurbiprofen (Flurizan or MPC-7869). Cyclophosphamide is a potent anti-inflammatory and immunomodulatory drug acting primarily by inhibiting the proliferation of immune cells.104 Excess tumour necrosis factor-alpha (TNF-α) has been shown to mediate the disruption in synaptic memory mechanisms caused by Aβ in addition to its proinflammatory functions.105 Etanercept, an antagonist of TNF-α, delivered by perispinal administration in AD patients, showed great potential in a pilot study. Among traditional medicine products, resveratrol, a component of grapes, berries and other fruits, is a polyphenol that has been shown to mediate its effects through modulation of many different pathways. For instance, resveratrol has been shown to reduce the expression of inflammatory biomarkers and induce antioxidant enzymes.106 Lastly, curcumin, a polyphenolic molecule safely used as a food colouring, proved to be immunomodulatory and has shown Aβ-40 aggregation inhibition properties in vitro and in vivo.107
Glutamate-Mediated Neurotoxicity
The glutamatergic system has long been recognized for its role in learning and memory and recent studies indicate the involvement of glutamate-mediated neurotoxicity in the pathogenesis of AD.108, 109 The neurotransmitter glutamate activates several classes of receptors and especially three major types of ionotropic receptors: α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), kainate and N
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


