The adrenal gland produces glucocorticoids, mineralocorticoids, sex hormones, and catecholamines. Deficiency or excess of any of these hormones results in clinical disease. Aldosterone, a mineralocorticoid that regulates sodium and water balance, is produced in the outermost of three layers that comprise the adrenal cortex, the zona glomerulosa. Cortisol, a glucocorticoid that regulates numerous metabolic processes, is produced in the middle layer of the adrenal cortex, the zona fasciculata. The innermost layer of the adrenal cortex, the zona reticularis, is primarily responsible for synthesizing and secreting androgenic steroid hormones that regulate sexual development. The central region of the adrenal gland, the medulla, produces catecholamines, which stimulate the sympathetic nervous system.
Adrenocortical insufficiency
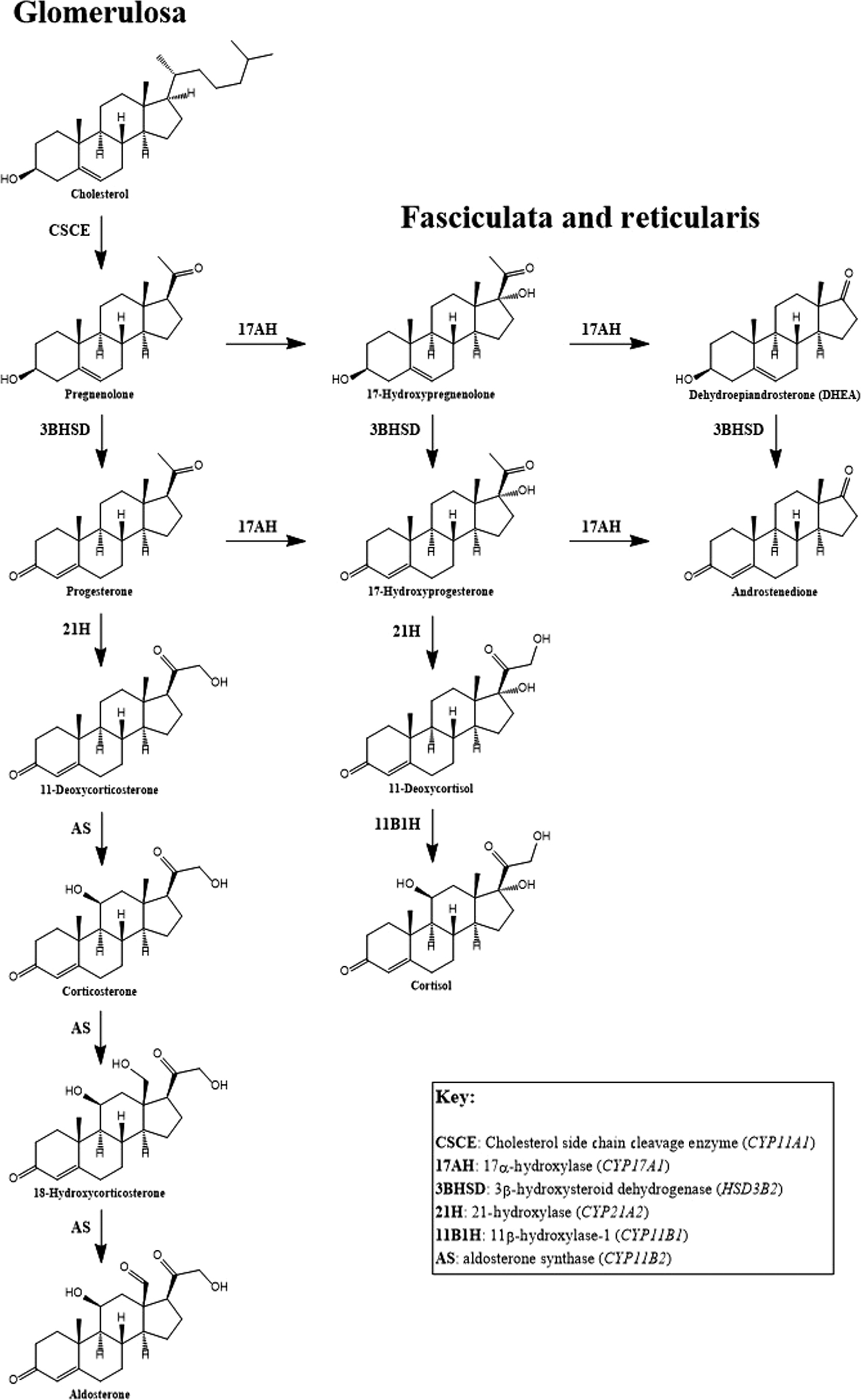
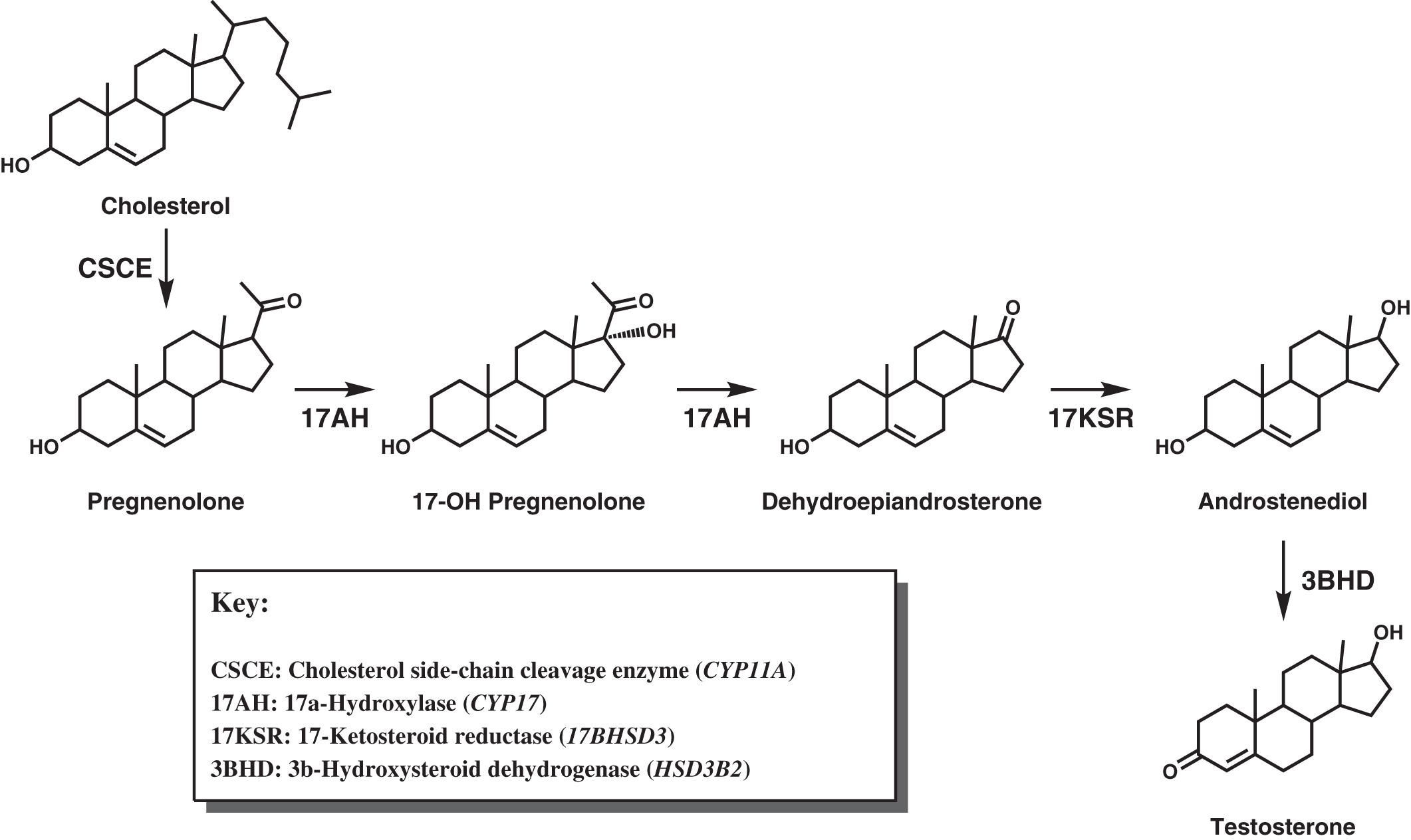
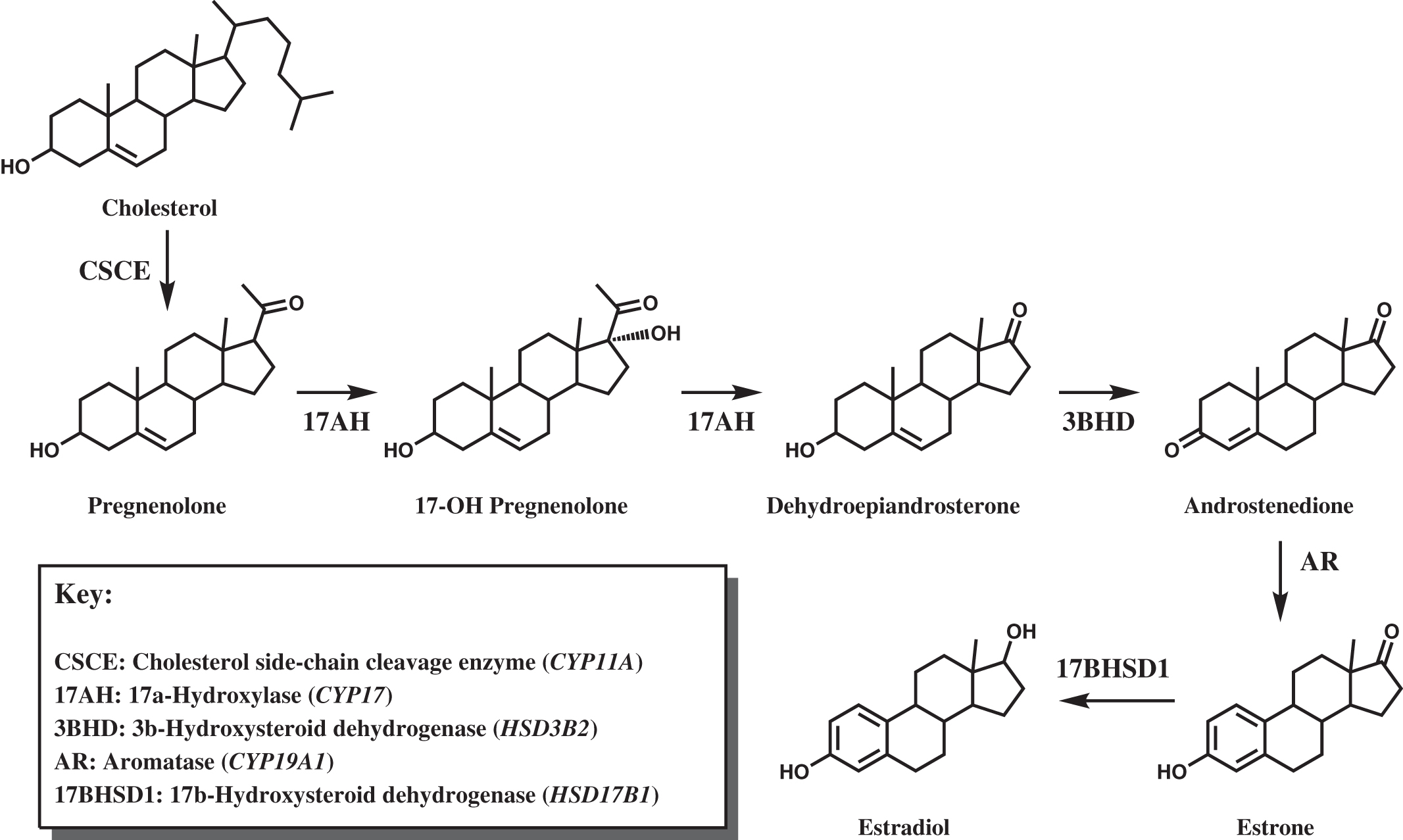
Deficiency of adrenocortical hormones causes serious, and often life-threatening, disease. Cortisol stimulates glucose production through hepatic gluconeogenesis and inhibits the action of insulin, resulting in hyperglycemia. Cortisol also helps maintain vascular tone and lysosomal integrity in cells, acting as an antistress or antishock hormone. Aldosterone promotes sodium reabsorption and potassium and hydrogen ion excretion by the renal tubules. The adrenal cortex shares with the gonads certain metabolic pathways for androgenic steroid hormone biosynthesis. The adrenocortical biosynthetic pathways to aldosterone, cortisol, and the adrenal androgens are shown in Fig. 4.1 . The Leydig cells of the testes synthesize testosterone from cholesterol by way of the 17α-hydroxylase pathway to dehydroepiandrosterone (DHEA), the same as in the adrenal cortex. ( Fig. 4.2 shows the predominant pathway to testosterone.). In the ovarian thecal cells, cholesterol is converted to androstenedione, which is taken up by the granulosa cells where it is converted to estrone and then estradiol ( Fig. 4.3 ). Adrenocortical failure can be manifested in cortisol deficiency, aldosterone deficiency, or combined deficiencies of both hormones. Combined adrenocortical failure is the clinical definition of Addison disease .
Detecting cortisol deficiency
Circulating cortisol concentrations are influenced by the presence of cortisol-binding globulin (CBG), also known as transcortin or serpin A6, an α-globulin encoded by the SERPINA6 gene . If hepatic synthesis of CBG is deficient due to malnutrition, malabsorption, or liver disease, or from protein loss due to nephrotic syndrome, total cortisol concentrations may decline below the reference interval, yet the biologically active free cortisol concentration is adequate. Conversely, elevated CBG concentrations can raise total cortisol levels without increasing free cortisol activity . Normally, about 70% of circulating cortisol is bound to CBG, 20% is bound to albumin, and 10% of total cortisol is unbound and biologically active . Many hormones are highly protein-bound, either to specific hormone-binding proteins or albumin, or most commonly, a combination of both. The proteins buffer the concentration of free, active hormone, preventing abrupt changes in hormonal activity that could have unpleasant, or even life-threatening physiological consequences.
There is great interest in measuring free cortisol . In intensive care settings, where patients are frequently malnourished, low CBG has the potential to prompt the overdiagnosis of Addison disease . Measuring CBG allows calculation of a free cortisol index, but there is disagreement over the reliability of calculated free cortisol estimates . Direct measurement of free cortisol concentration by equilibrium dialysis or ultrafiltration is cumbersome and rarely done. However, two alternatives exist for estimating the free cortisol concentration: salivary cortisol and urinary free cortisol (UFC).
Salivary cortisol reflects free cortisol concentrations in the blood . The saliva specimen is typically collected on a sponge and expressed into a test tube before filtration and analysis by either immunoassay or mass spectrometry . In addition to cortisol, there is interest in measuring other steroids in saliva. A recent report describes the measurement of classical and 11-oxygenated androgens in human saliva using liquid chromatography-tandem mass spectrometry (LC-MS/MS) . Steroid profiling by LC-MS/MS, at one time limited mostly to laboratories testing athletes for performance-enhancing drugs, is becoming increasingly common for clinical laboratory assessment of endocrine disorders.
Clinical symptoms and signs of adrenocortical insufficiency
Deficient cortisol production causes vascular instability, hypotension, and hypoglycemia, especially at times of severe stress . Patients feel weak with general malaise, loss of energy, and may display nausea, vomiting, or diarrhea. Primary adrenocortical failure is named after the 19th century English physician Thomas Addison, who described the disease in 1855. The clinical features of Addison disease include weakness, lethargy, increased fatigability, anorexia, nausea, vomiting, diarrhea (sometimes alternating with constipation), weight loss, abdominal pain, salt craving, muscle and joint pain, and postural dizziness. The clinical signs of Addison disease are mottled and increased skin pigmentation, decreased hair growth, hypotension, dehydration, tachycardia, and pallor.
Laboratory findings in primary adrenal insufficiency include hypoglycemia, a normocytic normochromic anemia, modest eosinophilia, and relative lymphocytosis. Hyponatremia, hyperkalemia, hyperchloremia, decreased bicarbonate, mild systemic acidosis, relatively alkaline urine, and azotemia result from mineralocorticoid deficiency. Mineralocorticoid deficiency produces type IV renal tubular acidosis (RTA) when the failure to excrete hydrogen ions leads to systemic acidosis. The failure to excrete potassium leads to hyperkalemia, whereas enhanced loss of sodium produces hyponatremia. In contrast, types I and II RTA are characterized by hypokalemia due to potassium loss in the urine. RTA type I results from defective secretion of hydrogen ions in the distal convoluted tubule, which impairs the production of bicarbonate. RTA type II is caused by defective reabsorption of bicarbonate ions in the proximal convoluted tubule, leading to excessive bicarbonate loss in the urine. RTA type III is a combination of proximal and distal RTA. When bicarbonate is either not produced (RTA type II) or is lost in the urine (RTA types I and IV), sodium is reabsorbed in an attempt to reverse hyponatremia, which elevates the cotransported chloride. Thus as bicarbonate declines and chloride rises, the patient experiences a hyperchloremic, normal anion gap acidosis. Relative or absolute hyperchloremia may be masked by electrolyte depletion and hyponatremia. Psychiatric symptoms of organic brain syndrome (impaired memory, confusion), depression, and psychosis may affect up to 40% of RTA patients.
In primary adrenal insufficiency, hyperpigmentation is a result of chronically elevated adrenocorticotropic hormone (ACTH) concentrations. Hyperpigmentation can also be observed in ectopic ACTH secretion causing Cushing syndrome, the POEMS syndrome (peripheral neuropathy, organomegaly, endocrine dysfunction, monoclonal gammopathy, and skin pigmentation), and uncommonly in Cushing disease. ACTH shares its N-terminal 13 amino acid sequence with one of the melanocyte-stimulating hormones, α-MSH, giving ACTH intrinsic melanotropic activity. In the intermediate lobe of the fetal pituitary gland, ACTH is enzymatically cleaved by prohormone convertase 2 to α-MSH and corticotropin-like intermediate lobe peptide. The intermediate lobe of the pituitary is not present in adults.
Patients with secondary or tertiary adrenal insufficiency from, respectively, ACTH or corticotropin-releasing hormone (CRH) deficiency, do not exhibit hyperpigmentation. Also, patients solely deficient in ACTH or CRH do not manifest severe dehydration, hypotension, hyperkalemia, or acidosis because aldosterone production and release are primarily regulated by angiotensin II; ACTH is only a minor regulator of mineralocorticoid production and release. Symptoms associated with ACTH or CRH deficiency are usually mild compared with Addison disease (primary adrenocortical failure) and may display hyponatremia, but not hyperkalemia. Hyponatremia can develop in the setting of cortisol deficiency because of a reduced ability to excrete a free water load.
Inability to increase glucocorticoid or mineralocorticoid production in response to acute stress precipitates “Addisonian crisis,” in which patients present with profound shock, hypotension, hypoglycemia, acidosis, hyponatremia, and hyperkalemia . Addisonian crisis is much more typical of primary adrenal failure than isolated ACTH deficiency. Untreated, Addisonian crisis can be fatal.
Causes of adrenocortical insufficiency
A list of some of the causes of adrenocortical insufficiency appears in Table 4.1 . Autoimmune destruction of the adrenal gland is the most common cause of primary adrenocortical failure in the industrialized world . Less commonly, the adrenal cortex can be destroyed by hemorrhage, infection [tuberculosis (TB) is the most common], trauma, adrenalectomy, and neoplastic disease. Adrenal dysgenesis, inborn errors of adrenocortical steroidogenesis, adrenoleukodystrophy (ALD), familial glucocorticoid deficiency, congenital adrenocortical hypoplasia, and certain drugs (e.g., the blockers of steroid synthesis: mitotane, aminoglutethimide, trilostane, ketoconazole, metyrapone, and the glucocorticoid receptor (GR) blocker RU-486), are rare causes of adrenocortical insufficiency. Familial glucocorticoid deficiency is a heterogeneous group of predominantly autosomal recessive disorders (but X-linked inheritance has also been described), with defects in the ACTH receptor gene located on chromosome 18p11.2 . Degeneration of the fasciculata and reticularis layers of the adrenal cortex is observed. A Reye-like syndrome involving hepatic encephalopathy may also be observed .
| Endogenous causes |
| Autoimmune polyglandular syndrome types 1 and 2 |
| Congenital adrenal hyperplasia |
| Congenital adrenal hypoplasia |
| Adrenoleukodystrophy |
| Intra-adrenal hemorrhage |
| Neoplastic infiltration |
| Exogenous causes |
| Infection (granulomatous disease, TB, sarcoidosis, histoplasmosis, blastomycosis, sporotrichosis, cryptococcosis, coccidioidomycosis) |
| Drugs that block steroid synthesis |
| Etomidate (inhibits 11β-hydroxylase) |
| Mitotane (inhibits cholesterol side-chain cleavage enzyme, 11β-hydroxylase, 18-hydroxylase, and 3β-hydroxysteroid dehydrogenase) |
| Aminoglutethimide (inhibits cholesterol side-chain cleavage enzyme and aromatase) |
| Ketoconazole (inhibits cholesterol side-chain cleavage enzyme, 17α-hydroxylase, 17,20-lyase, and 11β-hydroxylase) |
| Metyrapone (inhibits 11β-hydroxylase) |
| Benzodiazepines (inhibit 17- and 21-hydroxylase, and possibly 11β-hydroxylase) |
| Osilodrostat (inhibits aldosterone synthase and 11-β-hydroxylase) |
ACTH deficiency, causing secondary adrenocortical insufficiency, can result from suppression of the hypothalamic-pituitary-adrenal (HPA) axis with exogenous glucocorticoids, destructive or compressive pituitary tumors and their treatment, hypothalamic tumors and their treatment (leading to CRH deficiency), cerebral trauma, irradiation, infection, bleeding, congenital malformations of the pituitary gland or hypothalamus, and idiopathic hypopituitarism. Several transcription factor mutations are now recognized as causes of ACTH deficiency . In some cases, there are multiple anterior pituitary hormone deficiencies. Molecular testing is available for POU1F1 (Pit-1) [cytogenetic location 3p11.2, codes for POU transcription factor 1 (Pit1, growth hormone factor 1)] and PROP 1 (location 5q35.3, codes for paired like homeodomain factor 1) mutations.
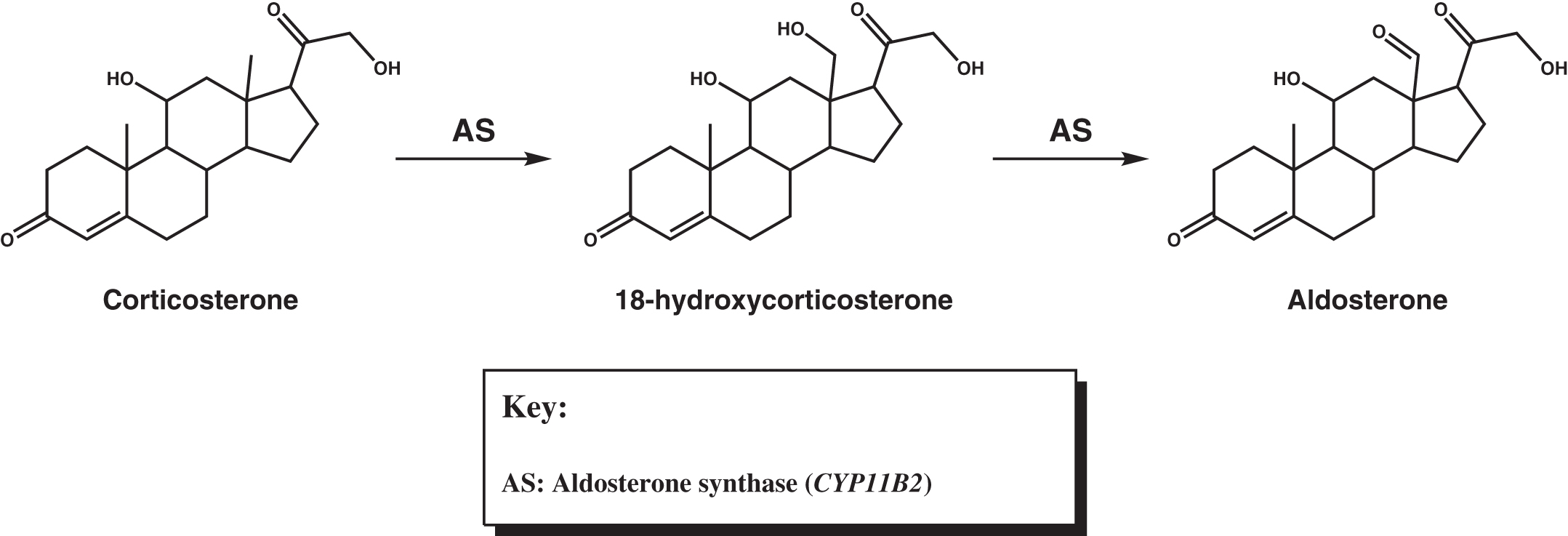
Isolated mineralocorticoid deficiency is rare, although during the development of Addison disease symptoms of mineralocorticoid deficiency can be seen before glucocorticoid deficiency becomes evident. The most common cause of isolated mineralocorticoid deficiency is inadequate renin production due to interstitial nephropathy or diabetic nephropathy. Rarely, aldosterone deficiency results from deficiency in aldosterone synthase ( CYP11B2 ) . This enzyme converts corticosterone into aldosterone via 18-hydroxylation to 18-hydroxycorticosterone, followed by 18-oxidation to aldosterone ( Fig. 4.4 ). Patients with primary adrenocortical hypofunction usually experience cortisol deficiency or combined deficiencies of cortisol and aldosterone (primary adrenocortical failure). In patients with primary adrenocortical failure, glucocorticoid deficiency may precede mineralocorticoid deficiency (or rarely, vice versa). Isolated cortisol deficiency results from insufficient ACTH (secondary adrenocortical insufficiency) or CRH (tertiary adrenocortical insufficiency).
Resistance to glucocorticoids and mineralocorticoids has been reported . Glucocorticoid resistance syndrome (GRS) is caused by loss of function mutations in the NR3C1 gene, located on chromosome 5q31, which encodes the glucocorticoid receptor (GR). Similar to other forms of primary adrenal insufficiency, ACTH is elevated in GRS with consequently increased levels of adrenal androgens causing hirsutism, oligomenorrhea, infertility, and precocious puberty. Elevated ACTH also leads to 11-deoxycortisol (11-DOC) overproduction, causing hypokalemic hypertension. It is unclear whether increased circulating cortisol concentrations totally compensate for glucocorticoid resistance. In addition to elevations in ACTH and cortisol, there is increased UFC excretion and increased urinary excretion of 17-hydroxycorticosteroids.
Mutations in the mineralocorticoid receptor gene, NR3C2 , located on chromosome 4q31, or one of the three genes encoding the subunits of the epithelial sodium channel ENaC ( SCNN1A , SCNN1B , and SCNN1G ), located on chromosome 12p13.31, cause pseudohypoaldosteronism (PHA) Type 1 . There are two subtypes of PHA Type 1: In the autosomal dominant form, associated with mutations in the NR3C2 gene, the kidney is selectively resistant and the clinical presentation is of early-onset salt-wasting that resolves within a few years of birth. More severe is the autosomal recessive, multiorgan (or generalized) form of PHA type 1, caused by ENaC mutations, that involves the sweat glands, the salivary glands, and colon in addition to the kidney. This disorder does not improve with advancing age. PHA type 2 does not cause salt-wasting.
Although not usually predisposing to Addisonian crisis from severe adrenal suppression, nasal steroids can nevertheless affect the HPA axis . Symptomatic adrenal insufficiency from withdrawal of inhaled steroids in asthma has been reported as well as adrenal crisis in children receiving high doses of inhaled steroids . Intracranial hypertension can also result from inhaled steroids . Bone density is another safety concern when high-dose inhaled steroids are used in children , but not in adults . Kannisto et al. reported that 25% of children treated with inhaled steroids displayed mild adrenal insufficiency when challenged with a 0.5 μg/1.73 m 2 dose of ACTH . The inhaled glucocorticoids fluticasone and ciclesonide, compared with beclomethasone and budesonide, may produce less adrenal suppression .
Laboratory investigation of adrenocortical insufficiency
Plasma cortisol levels of <5 μg/dL in the morning or during times of stress are presumptive evidence of glucocorticoid (cortisol) deficiency. Levels ≥18–20 μg/dL essentially exclude cortisol deficiency. If Addison disease is suspected and the patient is in crisis, a plasma cortisol level should be requested and the patient should be administered saline and glucocorticoids in pharmacologic doses (4 mg dexamethasone per m 2 of body surface area; dexamethasone is preferred because it has higher glucocorticoid activity than cortisol and does not interfere with cortisol measurements) . The patient should be treated for presumed Addisonian crisis until the cortisol concentration is determined, but the cortisol level is not urgent because dexamethasone treatment has little short-term risk. ACTH can be measured at a later time if desired to confirm the diagnosis of primary adrenocortical insufficiency. A flow diagram for the investigation of suspected glucocorticoid deficiency appears in Fig. 4.5 .

Patients presenting with Addisonian crisis
When a patient presents with hypotension, hypoglycemia, hyperpigmentation, hyponatremia, hyperkalemia, and systemic acidosis that are not explained by history, the diagnosis of Addison disease (primary adrenocortical failure) is strongly suggested. The diagnosis is confirmed by a plasma cortisol concentration of <5 μg/dL at the time of stress. A concomitant elevation in plasma ACTH supports the diagnosis of Addison disease. Additional confirmation of the diagnosis can be obtained with a 1-h ACTH stimulation test, or an insulin tolerance test (ITT). In the ITT, insulin is administered to induce hypoglycemia, which is a powerful central stimulant for the release of CRH, inducing ACTH and subsequent cortisol secretion. Some authorities state that the ITT is indicated only when recent ACTH or CRH deficiency is suspected, and the 1-h ACTH stimulation test could be falsely normal. Otherwise, the 1-h ACTH stimulation test provides similar information to the ITT. In patients with a clinical history that suggests potential ACTH deficiency, the ITT can be performed to concurrently evaluate the cortisol and growth hormone (GH) response to hypoglycemia. The 1-h ACTH stimulation test is much easier, safer, and cheaper to perform than the ITT. Because neither of these tests differentiates primary adrenocortical failure from ACTH deficiency, a 2-day (or longer) ACTH stimulation test can be performed instead. Despite prolonged stimulation from exogenous ACTH in the 2-day or longer ACTH stimulation test, in Addison disease the cortisol levels remain <5 μg/dL, confirming primary adrenocortical failure.
In the 1-h ACTH stimulation test, the traditional dose is 250 μg. In multiday ACTH stimulation tests, the dose is 250 μg/day. Because 250 μg of ACTH is a suprapharmacologic dose, investigators have studied the usefulness of giving only 1 μg in hopes of detecting subtler degrees of adrenal insufficiency . Indeed, there are patients who have normal responses to 250 μg of ACTH yet inadequate responses to 1 μg of ACTH. This remains a point of controversy, and the 1 μg ACTH dose has not been accepted as universally superior to the 250-μg dose, particularly in children . A lower cortisol response to 250 μg of ACTH may not predict better outcomes in patients with septic shock who are treated with glucocorticoids . An ACTH dose of 0.5 μg has been investigated in premature infants . Very preterm infants requiring ventilation had lower cortisol responses to stimulation than less ill, nonventilated, preterm infants.
The need to rapidly detect glucocorticoid deficiency remains clinically important in the intensive care setting where the use of glucocorticoids in sepsis remains controversial . If the patient can be shown to be glucocorticoid deficient, administration of glucocorticoids is justified.
Patients with suspected glucocorticoid deficiency who do not present acutely ill
If a patient presents with signs or symptoms suggestive of, but not diagnostic for, glucocorticoid deficiency, a laboratory investigation should be pursued before glucocorticoid replacement therapy is initiated. A patient ill enough to require acute glucocorticoid therapy should be evaluated the same as patients in Addisonian crisis.
Fasting plasma cortisol levels should be obtained on at least two separate days between 0600 and 0800 h, preferably at the same time each day. Stress or morning fasting cortisol concentrations <5 μg/dL are presumptive evidence of glucocorticoid deficiency ( Fig. 4.5 ). Stress or morning fasting cortisol concentrations 5–18 μg/dL are potentially abnormal. Stress or morning fasting cortisol values ≥18–20 μg/dL essentially exclude glucocorticoid insufficiency, and further workup is not usually indicated. However, if a stress or morning cortisol less than 18–20 μg/dL raises the suspicion of glucocorticoid deficiency, a 1-h ACTH stimulation test or an ITT should be performed. Following ACTH stimulation, a peak cortisol of <18–20 μg/dL and a change in cortisol over baseline of <7–10 μg/dL (Δcortisol) are abnormal and are diagnostic for glucocorticoid deficiency. Further evaluation may or may not be indicated depending upon the clinical circumstances. For example, if the ACTH level is elevated at the time that the cortisol is low, primary adrenocortical insufficiency is highly likely, and further testing usually is not needed. If adrenal autoantibodies are detected, and the finding is associated with low stress or morning cortisol levels and an abnormal 1-h ACTH stimulation test or ITT, further testing should not be pursued because these results confirm autoimmune Addison disease.
Once a patient has been confirmed as having glucocorticoid deficiency, the multiday ACTH stimulation test is useful for differentiating primary glucocorticoid deficiency (primary adrenocortical failure, Addison disease) from secondary (or central) glucocorticoid deficiency due to inadequate ACTH. When the adrenal gland is exposed to sustained exogenous ACTH stimulation, in cases of endogenous ACTH deficiency, the previously atrophied adrenal gland will begin to produce cortisol. However, in primary adrenocortical insufficiency, even with supraphysiologic exogenous ACTH stimulation, the adrenal gland is unable to increase cortisol production. Thus an abnormal multiday ACTH stimulation test confirms primary glucocorticoid deficiency because there is a deficient cortisol response to ACTH even when maximal doses of ACTH are applied.
In cases of Addison disease or ACTH deficiency, further confirmation of a defect in the HPA axis can be achieved by a metyrapone (Metopirone) stimulation test. Metyrapone is a pyridine derivative that inhibits 11β-hydroxylase, the enzyme that converts 11-deoxycortisol to cortisol in the zona glomerulosa of the adrenal cortex (see Fig. 4.1 ). Hence, metyrapone inhibits cortisol production. A typical protocol involves administering 30 mg metyrapone per kg of body weight at midnight and then measurement of plasma cortisol, ACTH, and 11-deoxycortisol at 0800 the next morning. A normal response is a decrease in cortisol, and an increase in ACTH (>150 pg/mL) and 11-deoxycortisol (>7 μg/dL). Some investigators believe that metyrapone testing is the most sensitive approach to determining adrenal insufficiency . Patients may have normal basal concentrations of cortisol and ACTH, normal responses to ACTH stimulation, and normal responses to insulin-induced hypoglycemia, yet have deficient responses to metyrapone. Presently, metyrapone is not available for clinical use in the United States.
Measuring morning ACTH levels may also assist in differentiating primary from secondary glucocorticoid deficiency. In primary glucocorticoid deficiency, ACTH is elevated, whereas in secondary glucocorticoid deficiency, ACTH is either low or inappropriately normal (when cortisol is low). However, ACTH is labile, and its concentration is highly variable over the course of a day because of the normal diurnal variation that is observed in the operation of the HPA axis. Therefore ACTH is not as reliable a marker for glucocorticoid deficiency as, for example, thyrotropin (TSH) is for hypothyroidism.
ACTH deficiency is diagnosed when the multiday ACTH stimulation test is normal, yet the 1-h ACTH stimulation test or ITT is abnormal. ACTH deficiency can be primary (due to pituitary disease) or secondary (due to hypothalamic disease). As noted earlier, an inappropriately normal or low concentration of ACTH concurrent with a depressed cortisol level in the morning or at a time of stress also supports the diagnosis of ACTH deficiency. Because the differentiation between hypothalamic and pituitary disease is usually accomplished radiographically, the CRH test is only rarely used to differentiate primary from secondary ACTH deficiency.
If primary glucocorticoid deficiency (primary adrenocortical failure) is confirmed, evaluation of mineralocorticoid status is indicated. Minimal evaluations include serum electrolytes and plasma renin concentration or activity (plasma renin activity; PRA) to define whether there is: (1) normal mineralocorticoid function (normal electrolytes, normal renin), (2) compensated mineralocorticoid deficiency (normal electrolytes, elevated renin), or (3) uncompensated mineralocorticoid deficiency (hyponatremia, hyperkalemia, and elevated renin).
Tests of adrenal function
Adrenocorticotropic hormone stimulation tests
The 1-h ACTH stimulation test determines whether or not the adrenal cortex can respond to a single, acute pharmacologic dose of ACTH with an increase in cortisol production. Although pig-derived corticotropin (ACTH) is available, it is not FDA-approved for diagnostic use. For diagnostic testing, a synthetic oligopeptide containing the active N-terminal 24 amino acids of corticotropin (ACTH has 39 amino acids) is used. The 24-amino acid analog is tetracosactide, also known as cosyntropin and by the trade names Cortrosyn (Amphastar Pharmaceuticals Rancho Cucamonga, CA), and Synacthen (Mallinckrodt, Staines-upon-Thames, United Kingdom—not available in the United States). Cosyntropin is available in generic form, as well. If there is primary gland failure or gland atrophy from ACTH deficiency, peak cortisol following a 250 μg dose of Cortrosyn is typically <18–20 μg/dL and the change from baseline is <7–10 μg/dL. The absolute cortisol level is a more sensitive measure of adrenocortical response than the change in cortisol from baseline.
The 1-μg Cortrosyn stimulation test has been proposed as an alternative to the 250-μg test because it is closer to physiological ACTH concentrations and may reveal a subtle degree of adrenal insufficiency not evident with the suprapharmacologic 250-μg Cortrosyn dose. However, a recent metaanalysis of studies comparing the two protocols concluded that both approaches have similar diagnostic accuracy . The detection of subtle cortisol deficiency is believed to be very important in the intensive care setting .
Prolonged Cortrosyn administration (8-h, 2-day, and 3- to 5-day ACTH stimulation tests) provides a higher level of sustained corticotropin stimulation to overcome adrenocortical atrophy that may have occurred from chronic ACTH deficiency. In primary adrenal failure, prolonged ACTH stimulation does not produce cortisol concentrations at or above the 18–20 μg/dL threshold regardless of the dose or duration of the Cortrosyn challenge. When hypocortisolism is the result of ACTH or CRH deficiency, prolonged and sustained exogenous cosyntropin will stimulate the atrophied gland to redevelop and produce a normal cortisol response. In acute ACTH or CRH deficiency, the serum cortisol response to stress is low whereas the response to exogenous cosyntropin can be normal. Several weeks or months of ACTH or CRH deficiency are required to induce atrophy of the adrenal cortex and a correspondingly deficient response to exogenous ACTH stimulation. If there is an abnormal serum cortisol level in response to stress and a normal Cortrosyn stimulation test when acute ACTH or CRH deficiency is a clinical possibility, an ITT or metyrapone test may be indicated. There is evidence that cosyntropin may interfere with some methods used to measure ACTH, although the stimulation test does not require measurement of ACTH.
The 1-h adrenocorticotropic hormone stimulation test
After a specimen is collected for baseline plasma cortisol, Cosyntropin (Cortrosyn) is administered intravenously or intramuscularly (25 units, or 250 μg) as a bolus. Cortisol is then measured in samples collected 30 and 60 min after administration. Some protocols specify collection of only one poststimulation sample at 45 min. If the adrenal cortex is functioning normally, the stimulated cortisol is >18–20 μg/dL, and the change from baseline is typically >7–10 μg/dL. Between the two criteria, the absolute cortisol level is more diagnostic. If the basal cortisol is >18–20 μg/dL, ACTH administration may produce little further elevation in serum cortisol because the patient may already be maximally stimulated at baseline.
Since plasma cortisol levels can be influenced by the concentration of CBG, it has been suggested that salivary cortisol after 250 μg cosyntropin stimulation may provide additional diagnostic value in patients predisposed to CBG alterations or patients with borderline plasma cortisol responses .
8-h, 2-day, and 3- to 5-day adrenocorticotropic hormone stimulation tests
When ACTH or CRH deficiency is suspected, and the physician wants to prove that the adrenal gland otherwise functions normally, a prolonged ACTH stimulation test may be performed. There are several variations intended to provide sufficient ACTH stimulation to reverse any atrophy that has developed from chronic ACTH or CRH inadequacy.
In the 8-h test, a baseline 24-h urine sample for UFC is measured. Assessment of UFC provides an integrated 24 h measurement of cortisol production. On day 2, a 24-h urine collection is begun, and Cortrosyn is delivered intravenously (25 units, or 250 μg) over 8 h. To demonstrate adequate adrenal function, the plasma cortisol at 60 min should be >18–20 μg/dL and at 8 h, >25 μg/dL, and UFC on day 2 should be approximately threefold greater compared with the baseline collection. With endogenous ACTH or CRH deficiency, the 8-h test may not be long enough to elicit a normal cortisol response. The 8-h ACTH stimulation test is rarely performed.
The 2-day and 3- to 5-day tests deliver Cortrosyn every day (an infusion over 8 h or via intramuscular injection or a continuous infusion of ACTH) with similar expected increases in cortisol and UFC. Presently, the 2-day test is the most popular prolonged ACTH stimulation test (1 day of baseline urine collection and 2 days of continuous ACTH administration). After maximal exogenous ACTH stimulation, if cortisol does not increase appropriately (>18–20 μg/dL), primary adrenal insufficiency is confirmed. Finding an elevated basal plasma ACTH concentration is consistent with a primary failure of cortisol production.
Corticotropin-releasing hormone stimulation test
In central glucocorticoid deficiency, ACTH versus CRH deficiency can be differentiated using the CRH stimulation test. In this test, if ACTH rises after CRH administration, glucocorticoid deficiency is due to CRH deficiency. However, if there is no increase in ACTH after CRH administration, there is evidence for pituitary failure to secrete ACTH. In the CRH stimulation test, ovine CRH (corticorelin ovine triflutate, trade name Acthrel; Ferring Pharmaceuticals, Parsippany, NJ) is administered by intravenous push in a dose of 1 μg/kg up to a maximum of 100 μg/dose . ACTH can be measured at baseline and at 15, 30, and 60 min. The 60-min sample is the most important observation.
Insulin-induced hypoglycemia test
Hypoglycemia is a strong stimulus of the HPA axis. In the insulin-induced hypoglycemia test (or ITT), 0.1 units of insulin per kg body weight are administered by intravenous push. Cortisol is measured at baseline and at 10, 20, 40, and 60 min. In obese individuals, or in individuals with other causes of suspected insulin resistance, the insulin dose should be increased to 0.15 U/kg.
The ITT is a potentially dangerous test that can induce severe hypoglycemia and seizures, especially in children who are GH and/or cortisol-deficient. The ITT must be supervised continuously by a physician or other health care provider who is trained in the recognition of hypoglycemia and its treatment. Adequate intravenous access must be maintained during the test, and the blood glucose must be measured with every blood draw. If the patient develops signs or symptoms of hypoglycemia during the test, blood glucose should be measured immediately with a bedside monitor. If the blood glucose is <60 mg/dL, a sample should be sent to the central laboratory for stat blood glucose measurement. If the blood glucose is confirmed to be <40 mg/dL, and hypoglycemic symptoms have not improved, the test should be terminated after a final sample for cortisol measurement is obtained. Intravenous glucose or glucagon (1 mg intravenously) can then be administered, and the patient can be fed.
If a child suffers a seizure during the ITT, the test should be immediately terminated with the intravenous administration of 1 mL/kg 50% dextrose (D50). Because of the high osmolality of D50, it is best administered rapidly into a large central vein or with great care more slowly into a smaller peripheral vein.
Glucagon stimulation test
Glucagon stimulates ACTH release from the pituitary, and the glucagon stimulation test (GST) has been proposed as an alternative to the ITT for diagnosis of GH deficiency and secondary adrenal insufficiency . Compared with the ITT, glucagon stimulation was equally sensitive for the detection of cortisol deficiency. The safety and simplicity of the GST is a considerable advantage over the ITT. In a typical protocol, 1–1.5 mg of glucagon is administered subcutaneously or intramuscularly. Peak plasma cortisol concentrations >18 μg/dL following glucagon stimulation are consistent with secondary adrenal insufficiency . The GST is most useful for assessing GH deficiency in adults.
Mineralocorticoid assessment
Aldosterone concentrations vary with salt intake and body position. Salt restriction can raise aldosterone levels approximately eightfold. To account for these variables, a 24-h urine sodium should be measured when plasma aldosterone is measured. Aldosterone levels can then be interpreted in relation to salt intake.
Prior to the measurement of basal plasma aldosterone concentrations, patients should receive their normal salt intake for 24–48 h, and be in the supine position for 30 min prior to collection of the blood specimen. Upright posture can double the plasma aldosterone concentration. Alternatively, a 24-h urine can be collected for aldosterone excretion to assess the daily aldosterone production. When aldosterone production and secretion are insufficient, normally there is an increase in plasma renin or PRA.
On normal sodium intake, aldosterone excretion is 3–10 μg/day. Values of 20–50 μg/day are achieved with a low-salt diet. If the diagnosis of mineralocorticoid deficiency is in question, plasma aldosterone and PRA should be obtained after 3 h of upright posture or after a day or more of salt restriction. Normally with such measures, aldosterone and PRA rise. Salt restriction should be undertaken with care so as not to precipitate Addisonian crisis. Low serum aldosterone, low urinary excretion of aldosterone, and elevated PRA are strong evidence of primary adrenal insufficiency.
If electrolytes are normal, these tests of mineralocorticoid function may be performed in patients being evaluated for primary adrenocortical failure. However, if the patient history suggests mineralocorticoid deficiency and includes hyponatremia, hyperkalemia, and a normal anion gap metabolic acidosis, there is no additional benefit from aldosterone measurement.
Aldosterone deficiency is treated with fludrocortisone (9α-fluorohydrocortisone, brand name Florinef, Monarch Pharmaceuticals, Inc., Bristol, TN). Patients with mineralocorticoid deficiency who are undertreated can display electrolyte abnormalities and hypertension from elevated renin and angiotensin II that cause vasoconstriction. In severe untreated mineralocorticoid deficiency, hypotension can quickly develop, followed by shock and/or death during times of severe stress.
Evaluation for specific causes of primary adrenocortical insufficiency
Autoimmune Addison disease
In the western world, autoimmune destruction of the adrenal glands is responsible for approximately 85% of Addison disease, excluding cases of congenital adrenal hyperplasia (CAH) . Prior to the discovery of antibiotics, Addison disease most commonly resulted from TB. The diagnosis of autoimmune Addison disease is established by identification of autoantibodies to 21-hydroxylase , typically measured using radioimmunoassay or chemiluminescent-labeled 21-hydroxylase and immunoprecipitation. An enzyme-linked immunosorbent assay (ELISA) has also been described for measuring 21-hydroxylase autoantibodies . Alternatively, the concentration of the 21-hydroxylase substrate, 17-OHP, can be measured, and results >1000 ng/dL are diagnostic for 21-hydroxylase deficiency, although the test does not discriminate between autoimmune and congenital causes .
Based on a European Expert Consensus Statement for diagnosis, treatment, and follow-up of primary adrenal insufficiency , when a patient with suspected primary adrenal insufficiency is negative for 21-hydroxylase autoantibodies, imaging studies are recommended, and if the patient is male, very long-chain fatty acids (VLCFA) to investigate the possibility of ALD. In children and adolescents presenting with primary adrenal insufficiency, the diagnosis of autoimmune polyendocrine syndrome type 1 (APS-1) should be considered. APS-1 is a rare autosomal recessive disorder presenting in childhood, caused by mutations in the autoimmune regulator (AIRE) gene on chromosome 21 . The AIRE encodes a transcription factor that appears to modulate self-antigen expression in the thymus, which is important during T-cell ontogeny. The diagnosis of APS-1 is based on the finding of two of the following three disorders: autoimmune Addison disease, mucocutaneous candidiasis, and hypoparathyroidism. Other frequent manifestations of APS-1 include enamel hypoplasia and enteropathy. Females with APS-1 often experience ovarian insufficiency. Less common complications include bilateral keratitis, periodic fever and rash, hepatitis, pneumonitis, nephritis, exocrine pancreatitis, and functional asplenia. Rarely, retinitis, metaphyseal dysplasia, red cell aplasia, and polyarthritis occur .
Autoimmune polyendocrine (or polyglandular) syndrome type 2 (APS-2) is a polygenic disorder that presents in later childhood or adulthood. APS-2 is far more common than APS-1 and occurs more frequently in women than in men. The diagnosis of APS-2 is based on the presence of two of three endocrinopathies: autoimmune Addison disease, type 1 diabetes, or autoimmune thyroid disease (AITD; Hashimoto thyroiditis, atrophic thyroiditis, or Graves disease). Serologic markers of type 1 diabetes include islet cell cytoplasmic autoantibodies, glutamic acid decarboxylase autoantibodies, IA-2 autoantibodies, and, in children, insulin autoantibodies. Markers of AITD are discussed in the chapter on thyroid disorders. The concurrence of Addison disease and AITD has been termed Schmidt syndrome , whereas the addition of type 1 diabetes identifies Carpenter syndrome . Because of the association of type 1 diabetes with human leukocyte antigen-DR3 and -DR4, APS-2 is associated with these same human leukocyte antigens .
Although adrenal autoantibodies have been reported in the syndrome of immunodysregulation, polyendocrinopathy, enteropathy, X-linked (IPEX), there are no reports of Addison disease in this disorder. IPEX is a rare, autosomal recessive disorder associated with mutations in the FoxP3 transcription factor, a protein that influences the development and function of regulatory T cells .
In asymptomatic subjects positive for adrenal cortex autoantibodies (ACA) and/or autoantibodies to 21-hydroxylase, yearly cosyntropin stimulation testing can be used to detect incipient autoimmune adrenal insufficiency. Deficient cortisol responses to cosyntropin predict the development of frank primary adrenal insufficiency in such autoantibody-positive individuals . In these cases, anticipatory, long-term hormone replacement can be provided to prevent the development of serious illness, such as Addisonian crisis.
Congenital adrenal hyperplasia
CAH is a group of autosomal recessive disorders caused by mutations in the genes encoding enzymes involved in the biosynthesis of cortisol. The most common cause of CAH (95% of cases) is a mutation in CYP21A2 , which codes for 21-hydroxylase, the enzyme that converts progesterone or 17-hydroxyprogesterone (17-OHP) to deoxycorticosterone (DOC) or 11-deoxycortisol (11-DOC), respectively (see Fig. 4.1 ). Deficiency in 21-hydroxylase is characterized by impaired adrenal production of aldosterone and cortisol. CYP21A2 overlaps another gene, TNXB , which encodes Tenascin-X, an extracellular matrix protein. Mutations affecting Tenascin-X cause an autosomal recessive form of Ehlers–Danlos Syndrome (EDS), and up to 10% of patients with salt-wasting CAH display some of the clinical features of EDS .
The remaining 5% of CAH cases are caused by deficient 11β-hydroxylase, 17-hydroxylase, 3β-hydroxysteroid dehydrogenase type 2, P45 oxidoreductase, and P450 cholesterol side-chain cleavage enzyme deficiencies, and all but 11β-hydroxylase deficiency are exceedingly rare. CAH is the most common cause of adrenocortical insufficiency in newborns. Rarely, adrenal failure occurs in newborns from traumatic adrenal hemorrhage or adrenal hemorrhage from sepsis. Because of the large fetal adrenal gland relative to body size, mechanical trauma to the gland is more likely at birth than at any other time during life.
Neonatal screening for 21-hydroxylase deficiency by measurement of its substrate, 17-OHP, is relatively common in developed countries, but has a low positive predictive value. Measurement of 17-OHP by LC-MS/MS decreases the rate of false-positive results . Measurement of 21-deoxycortisol, a side-product formed by hydroxylation of 17-OHP by 11β-hydroxylase, has been suggested as a better screening test because it is only formed in significant amounts when there is excess 17-OHP .
Older tests used to assess defects in adrenal steroid metabolism focused on urinary products of steroid metabolism; these include pregnanetriol, tetrahydrodeoxycortisol, 17-ketogenic steroids, 17-ketosteroids, and 17-hydroxycorticosteroids. All of these are hepatic metabolites of circulating steroids, but these tests are no longer in common use.
In response to cortisol deficiency, CAH patients overproduce ACTH, as well as the adrenal androgens DHEA and androstenedione (due to the accumulation of their precursors, 17-hydroxypregnenolone and 17-OHP). In females, overproduction of the adrenal androgens induces in utero virilization of the external genitalia, which can vary from mild fusion of the labia, to marked labial fusion with clitoromegaly, to complete masculinization with male external genitalia but with bilaterally undescended “testes,” a misnomer because they are in fact ovaries in their normal intra-abdominal location. Untreated, females will continue to virilize postnatally. The more profound the defect in cortisol biosynthesis, the greater the elevation in ACTH and the greater the degree of virilization in the female fetus. In males, adrenal androgen overproduction causes precocious puberty and some degree of hyperpigmentation of the male genitalia. As with virilization of females, the more profound the defect in cortisol biosynthesis, the greater the degree of precocious puberty in males .
In CAH, glucocorticoid insufficiency leads to malaise, failure to thrive, hypoglycemia, and vascular instability. In approximately one-half of infants affected with 21-hydroxylase deficiency, both cortisol and aldosterone production are impaired, the latter causing hyponatremia, hyperkalemia, acidosis, dehydration, and hypotension. In its most severe form, this type of “salt-wasting” 21-hydroxylase deficiency clinically presents at 10–14 days of age with Addisonian crisis. Because salt-wasting 21-hydroxylase deficiency is a more severe expression of 21-hydroxylase deficiency, it is associated with greater degrees of virilization in males and females. Untreated, mortality is very high in salt-wasting 21-hydroxylase deficiency CAH. The nonsalt-wasting form of the disease is referred to as “simple virilizing” 21-hydroxylase deficiency CAH.
In 11β-hydroxylase deficiency, the second most common cause of CAH, mineralocorticoid deficiency does not occur. Therefore although females with 11β-hydroxylase deficiency present with ambiguous genitalia, and males display precocious puberty, salt-wasting does not occur. Thus electrolyte and acid-base balance is normal, and Addisonian crisis does not develop in the newborn period or later. However, later in childhood, hypertension, with or without hypokalemia, can develop . This is discussed in more detail as follows.
The diagnosis of 21-hydroxylase and 11β-hydroxylase deficiencies depends upon elevated concentrations of 17-OHP in plasma or serum. Affected infants typically have random plasma 17-OHP concentrations >5000 ng/dL (>150 nmol/L; reference range <130 ng/dL, <4.2 nmol/L). 21-Hydroxylase deficiency and 11β-hydroxylase deficiency CAH can be differentiated by measuring 11-deoxycortisol. In 21-hydroxylase deficiency, both 11-deoxycortisol and cortisol are low, whereas in 11β-hydroxylase deficiency, deoxycortisol is elevated, and cortisol is low. Table 4.2 compares clinical and biochemical features of 21-hydroxylase and 11β-hydroxylase deficiency CAH.
| Enzyme deficiency | ||
|---|---|---|
| 21-Hydroxylase | 11β-Hydroxylase | |
| Percent of CAH cases | 90%–95% | ~5% |
| Gene | CYP21 | CYP11B1 |
| Plasma 17-OHP | Elevated | Elevated |
| Plasma 11-DOC | Depressed | Elevated |
| Plasma corticosterone | Depressed | Depressed |
| Plasma 11-deoxycortisol | Depressed | Elevated |
| Plasma cortisol a | Depressed | Depressed |
| Ambiguous genitalia | In females | In females |
| Precocious puberty | In males | In males |
| Salt loss | Approximately half of patients | Does not occur |
| Hypertension | No | Late childhood/adolescence |
a In mild forms of CAH, cortisol may be low-normal due to the compensatory increase in ACTH.
A “late-onset” or “attenuated” (nonclassical) form of 21-hydroxylase deficiency can present in female adolescents as hirsutism or virilization. Late-onset 21-hydroxylase-deficient CAH is much milder than the classical (“virilizing”) form of 21-hydroxylase deficiency of prenatal onset. Although baseline and post-ACTH stimulation 17-OHP levels in attenuated 21-hydroxylase-deficient CAH are higher than in controls, the values are not as high as those seen in classical 21-hydroxylase deficiency. Graphs are available in the literature that allow basal 17-OHP to be plotted against post-ACTH 17-OHP in diagnosing CAH gene carriers versus attenuated 21-hydroxylase deficiency versus classical 21-hydroxylase . In mild forms of CAH, cortisol may be low-normal from a compensatory elevation in ACTH. Gene carriers usually have normal basal levels, but elevated 17-OHP in response to ACTH stimulation.
21-Hydroxylase and 11β-hydroxylase deficiency CAH patients can be followed clinically during glucocorticoid replacement therapy by measuring an early morning 17-OHP or by measuring androstenedione at any time of the day . The treatment of both forms of CAH is cortisol replacement. In 21-hydroxylase deficiency CAH, aldosterone is replaced with fludrocortisone. With adequate glucocorticoid replacement, 17-OHP or androstenedione should not be elevated above the upper limit of the reference interval . Insufficient glucocorticoid replacement can allow excess adrenal androgen production and postnatal virilization, accelerated growth, premature closure of the epiphyses, and short stature . Overtreatment with glucocorticoids can produce poor growth from iatrogenic Cushing syndrome.
Hypertension, with or without hypokalemia, may also develop in children with 11β-hydroxylase deficiency . Hypokalemic hypertension results from elevated concentrations of 11-DOC, but the cause of 11-DOC elevations is not well understood. 11-DOC accumulation is not due to the inborn 11β-hydroxylase error since expression of the CYP11B1 mutation is limited to the zona fasciculata; CYP11B2 converts 11-DOC to corticosterone in the zona glomerulosa and is not expressed in the fasciculata. Theoretically, 11-DOC is elevated in cases of CYP11B1 mutations because of excessive ACTH stimulation of the glomerulosa; conversion of progesterone to 11-DOC by 21-hydroxylase is ACTH-dependent. 11-Deoxycortisol has neither substantial glucocorticoid nor mineralocorticoid activity, therefore elevations in 11-deoxycortisol cannot explain hypertension in 11β-hydroxylase deficiency.
A rare cause of CAH is a deficiency in 17α-hydroxylase, which converts pregnenolone to 17-hydroxypregnenolone and progesterone to 17-hydroxyprogesterone. In 17α-hydroxylase deficiency CAH, production of cortisol is deficient but mineralocorticoids are overproduced due to accumulation of pregnenolone, causing salt and water retention and consequently hypertension. Because the gonads share many steroidogenic pathways with the adrenal glands, males with 17α-hydroxylase deficiency fail to virilize because concurrent gonadal androgen production is insufficient. Deficient estrogen production impairs normal puberty in females with 17α-hydroxylase deficiency. Since both 11β-hydroxylase deficiency CAH and 17α-hydroxylase deficiency CAH can produce hypertension, their properties are compared in Table 4.3 .
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


