Introduction
A disorder of sexual development (DSD) can be defined as “…any problem noted at birth where the genitalia are atypical in relation to the chromosomes or gonads” . “Ambiguous genitalia” is a common term that is used to describe “atypical” internal or external genitalia. However, there are DSDs without ambiguity of the external genitalia that are not noted until later in life (e.g., pubertal failure). The definition of DSDs is not always hard and fast, or straightforward. For example, individuals with Turner syndrome are “female” yet Turner syndrome is a form of a DSD.
It is instructive to distinguish the terms “sex” and “gender.” Gender is defined culturally (e.g., “masculine,” “feminine,” etc.), whereas sex is defined biologically (e.g., male, female, or the individual does not fall into this binary classification because they have a DSD). While an individual’s karyotype is used to classify DSDs, the karyotype does not define one’s sex. Phenotypic anatomic findings (e.g., ovary/testis, clitoris/glans penis, labial majora/scrotum, labia minora/penile shaft, etc.) more properly define one’s sex (and when these features are not clearly male or female, a DSD exists). The expression of individual genes in the context of environmental influences (e.g., “endocrine disrupters”) ultimately determine one’s sex . Endocrine disruptors are environmental chemicals that may perturb the endocrine system. The most important gene in determining sex is the Y-chromosome SRY gene (see below) .
While a working knowledge of karyotypes is necessary for our discussion of DSDs, this is not meant to be a treatise on chromosomal analysis with regard to each of these disorders. Likewise, we will neither focus on the treatment of DSDs nor discuss the potential for fertility preservation in such conditions .
Male sex can be defined as the ability to produce sperm for reproduction. Female sex can be defined as the ability to produce ova for reproduction. Intersex is a controversial term that has been used on occasion (predominantly in the British literature) to describe individuals with ambiguous genitalia. Intersex is culturally a popular term that can allow for the inclusion of people with DSD into the sexuality and gender minorities abbreviated as lesbian, gay, bisexual, transgender, queer, and intersex (LGBTQI).
The term “intersexuality” is the condition of having both male and female characteristics (being intermediate between the sexes). Of note, the terms male pseudohermaphrodite, female pseudohermaphrodite, and true pseudohermaphrodite are archaic and are not used in modern medicine.
A DSD does not refer to a person’s sexual orientation. Sexual orientation can be defined as the sex of individuals to which one is sexually attracted . An individual’s internal conception of being male, female, both (nonbinary), or neither defines one’s “gender identity.” Sexual orientation and gender identity are different concepts. Gender identity is often appreciated prior to the recognition of one’s sexual orientation (e.g., children can perceive gender identity before they recognize their sexual orientation).
When gender identity is concordant with the sex assigned at birth, the individual can be defined as “cisgender.” When gender identity is discordant from sex assigned at birth, the individual can be described as “transgender.” As quoted directly from Shumer et al., “(a) transgender person feels a discrepancy between their sex assigned at birth and their gender identity” . Transgender is not a disease, illness, or a type of DSD. However, some transgender people will experience gender dysphoria, or a sense of extreme anxiety/distress over the social projections related to their sex assigned at birth . In this evolving field, the definition of gender dysphoria is controversial . Transgender individuals can be bisexual, lesbian, gay, etc. A biologic woman who has transitioned or is transitioning to a man is a transgender man or, more simply, a transmale. A biologic man who has transitioned or is transitioning to a woman is a transgender woman or, more simply, a transfemale. A person diagnosed with a DSD may also identify as transgender, and some studies have shown that there is an increased prevalence of a transgender identity in individuals with a DSD . The issues of transgender health care are further discussed in Chapter 17 .
For the purposes of this chapter, the terms “male” and “female” (and those related to sex, e.g., “boy” or “girl”) are used to reference the phenotypic sex of the individual, as phenotype relates to the DSD. Care providers treating these individuals should be acutely sensitive to the patient’s preferred pronouns and relation to their gender identity .
Genitalia: structure and development
Genitalia are the internal and external male or female reproductive organs. Developmentally, female and male genitalia are similar until 9 weeks. Their final form is observed at week 12 of development.
Ambiguous genitalia in individuals with the 46, XX karyotype may feature: an enlarged clitoris, urethral opening along, above, or below the clitoris, labia fusion, a labial mass (e.g., a gonad located in the labia), or an otherwise “male” phenotype with bilaterally undescended testes. Ambiguous genitalia in individuals with the 46, XY karyotype may feature: a small penis (<2–3 cm at birth), a urethral opening along, above, or below the penis, a pseudovagina, urogenital sinus or cloacal opening, small scrotum, or undescended testes. In the extreme, the external genitalia of an individual with a 46, XY karyotype can be completely female with an apparent clitoris, labia majora, labia minora, and vagina.
In utero , the products of genes on the Y chromosome and some autosomal gene products influence the undifferentiated (bipotential) gonad to develop into a testis ( Fig. 16.1 ). The most important Y-chromosome encoded gene is SRY (Sex Determining Region Y; chromosome Yp11.2). SRY encodes a high mobility group (HMG)-box family transcription factor. Its protein product, the testis-determining factor (TDF), initiates testes development. An important autosomal gene on chromosome 17q24.3 is SOX9 (SRY-Box 9), which is controlled by the TDF . The protein product of SOX9 serves as a transcriptional regulator. Together with steroidogenic factor 1 [SF-1, a.k.a. nuclear receptor subfamily 5 group A member 1 (NR5A1); chromosome 9q33.3], SOX9 regulates anti-Mullerian hormone [AMH, a.k.a. Mullerian-inhibiting hormone (MIH); 560 amino acids] gene expression ( AMH gene: chromosome 19p13.3; see below). Other genes involved in the testis-determining pathways include Six1, Six4, Map3k4, Gadd45g , and Hhat .
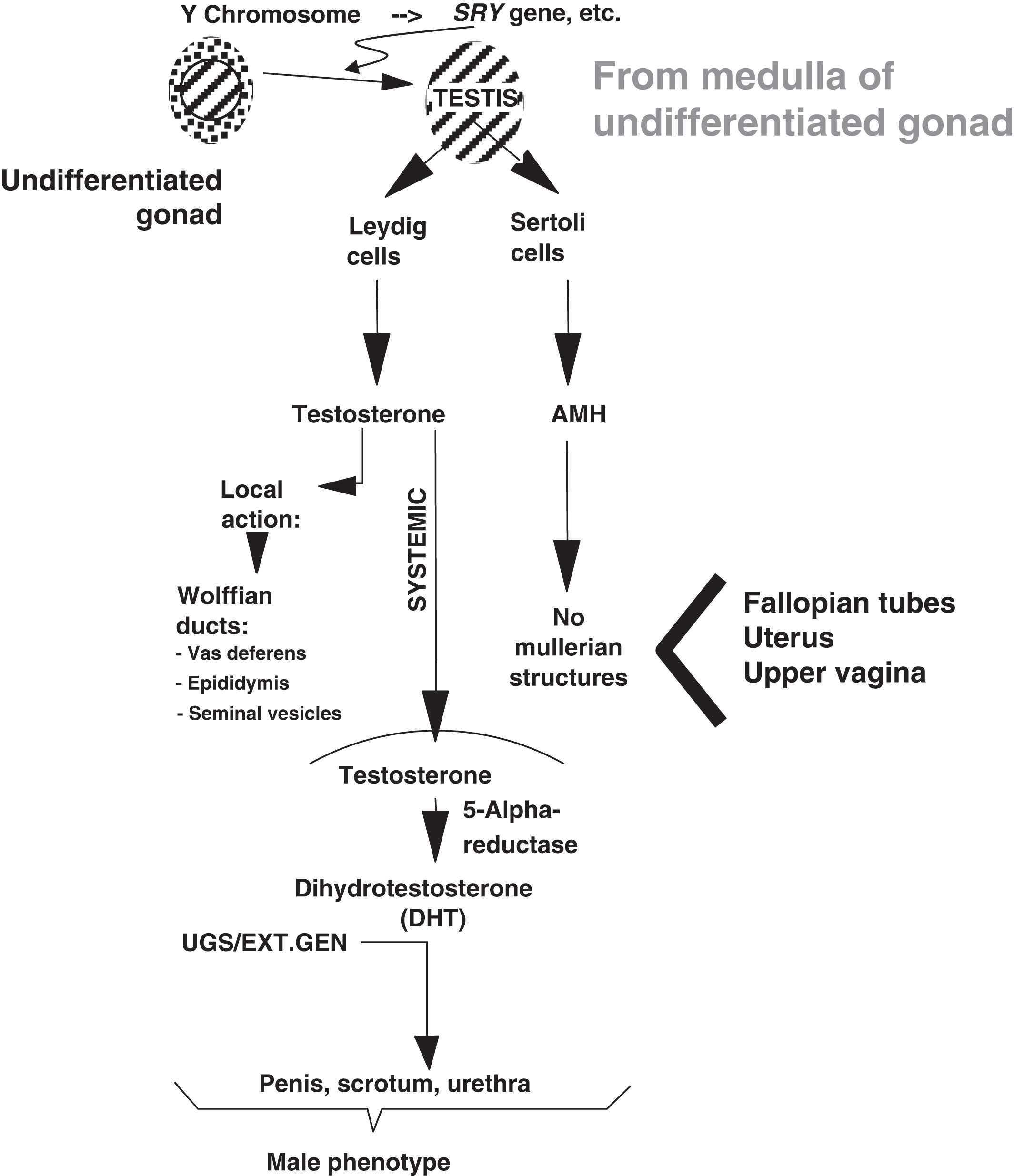
During development, Leydig cells produce testosterone that acts locally to induce the formation of the Wolffian ducts that include the vas deferens, epididymis, and seminal vesicles. Testosterone also circulates systemically and is converted into dihydrotestosterone (DHT) in target tissues. DHT developmentally induces the urogenital sinus and external genitalia to form the penis, scrotum, and male urethra. 5-alpha reductase is the enzyme that catalyzes the conversion of testosterone to DHT. In concert with the local and systemic effects of testosterone is the production of AMH from Sertoli cells, which inhibits the local formation in males of the Mullerian structures (i.e., fallopian tubes, uterus, and upper vagina).
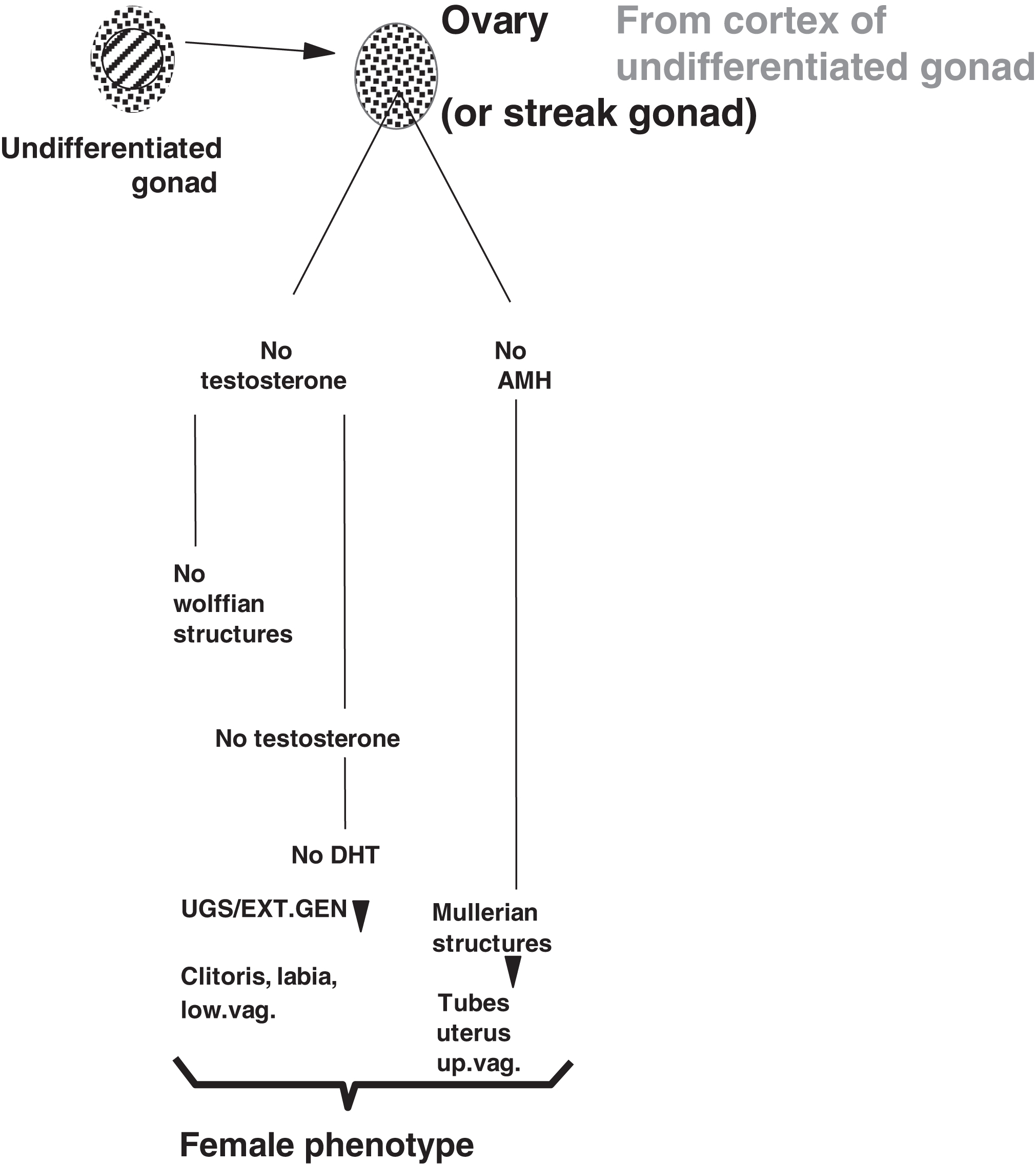
In the absence of a testis and local testosterone production, the Wolffian structures do not form ( Fig. 16.2 ). In the absence of circulating testosterone, DHT is not produced in the urogenital sinus and external genitalia leading to formation of the clitoris, labia, and lower vagina. In the absence of AMH, the Mullerian structures do form including the fallopian tubes, uterus, and upper vagina. The development of the external genitalia with reference to males and females is illustrated in Fig. 16.3 .
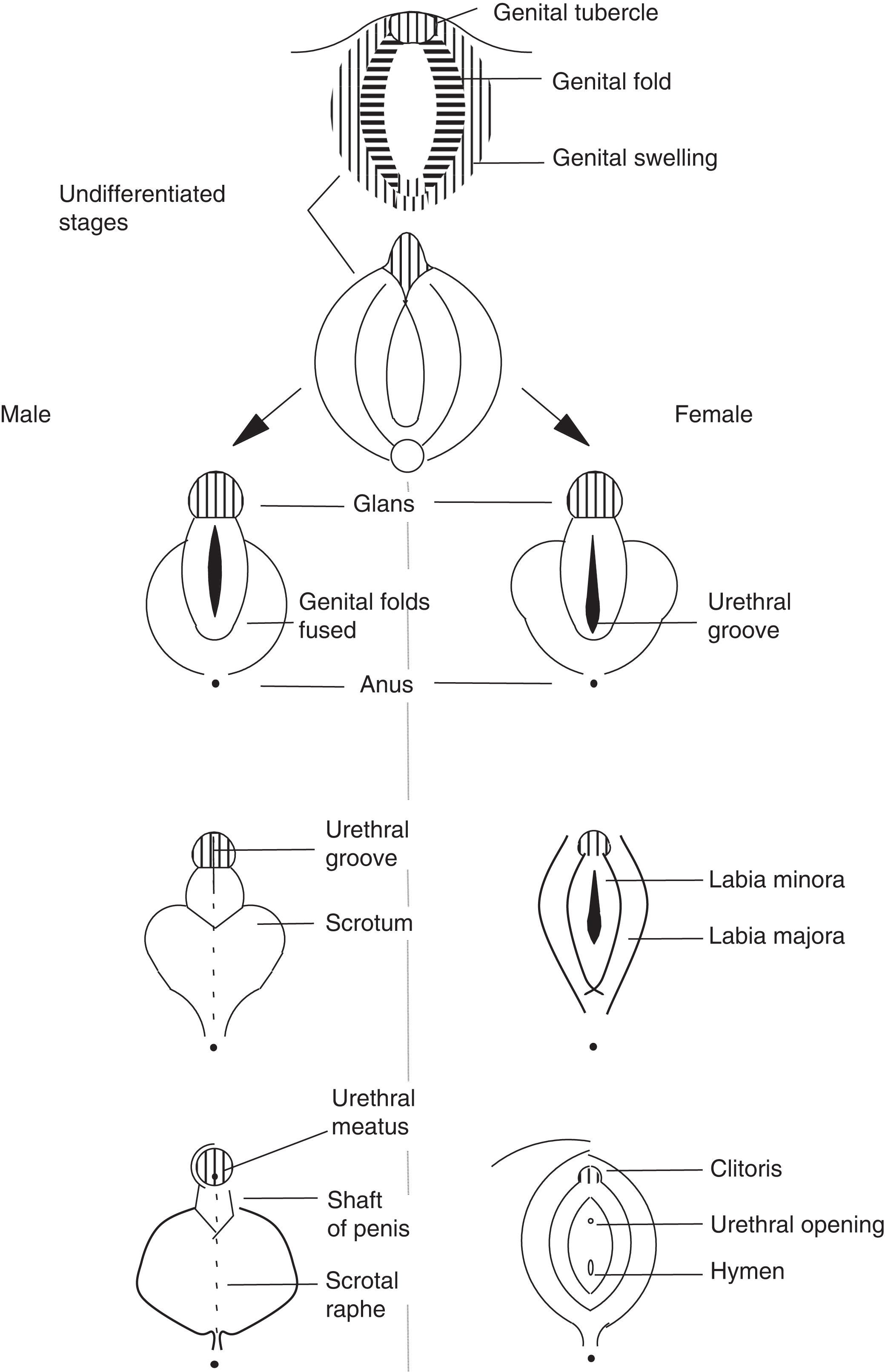
A clinical and laboratory approach to disorders of sexual development
In practical terms, commonly at the time of delivery or shortly thereafter, an individual with ambiguous genitalia is first recognized. The ambiguity of the visible (a.k.a. external) genitalia will trigger a clinical and laboratory evaluation that should be reasoned and methodical. It is certainly possible that a person can have a DSD that is not recognized early in life. Such is the case with complete androgen insensitivity syndrome (CAIS) where the “ambiguity” is not visible externally.
In order to understand the diagnostic evaluation of DSDs, we will review a system of classification of DSDs. Following the classification of the DSD, we will examine the biology and diagnosis of each DSD and the role played by the laboratory in the diagnosis of the DSD. We will then focus on DSDs presenting with ambiguous genitalia in order to provide a suggested conceptual, clinical, and laboratory approach to their evaluation.
Initial evaluation of persons with a disorder of sexual development
Currently, the karyotype of the individual guides the initial evaluation of the patient. Certainly, there can be other considerations [e.g., hyponatremia, hyperkalemia, acidosis, and/or hypoglycemia in cases of salt-wasting congenital adrenal hyperplasia (CAH)]. With modern cytogenetic laboratories, sex chromosome determination can be performed in a matter of days (48–72 h). Often, this can be followed within 24 h by reporting the fluorescence in situ hybridization (FISH) analysis for the SRY gene that indicates the presence of Y-chromosome material. In contrast, DSDs were historically classified according to gonadal histology, which is much more difficult to rapidly obtain.
DSDs can be classified as:
- 1.
Sex chromosome DSDs;
- 2.
46, XY DSDs; and
- 3.
46, XX DSDs.
In the text, the disorders will be sequentially discussed, in general, from (1) chromosomal errors, to (2) gonadal errors, to (3) biochemical errors in adrenal or placental steroid or sex steroid synthesis, to (4) target organ errors, and, lastly, to (5) malformations unrelated to known chromosomal, genetic, or biosynthetic errors.
The tables, however, summarizing characteristic features of 46, XY DSDs and 46, XX DSDs are ordered according to whether ambiguous genitalia are absent or present, and when ambiguous genitalia are absent, whether the karyotype and external genitalia are concordant (e.g., 46, XY with male external genitalia and 46, XX with female external genitalia) or discordant (e.g., 46, XY with female external genitalia and 46, XX with male external genitalia). This latter approach makes clinical sense: when the physician first encounters the affected patient, the patient’s physical examination will determine if ambiguous genitalia are present or absent. Next, the physician will “compare” the patient’s physical examination findings and the karyotype to help determine the choice of further evaluations that will likely include laboratory testing.
Sex chromosome disorders of sexual development
Table 16.1 lists the major sex chromosome DSDs. In these DSDs, sex chromosome anomalies are present and may cause the DSD.
| Anomaly | Comment |
|---|---|
| X-chromosome abnormalities |
|
| Y-chromosome abnormalities |
|
| X and Y chr. abnormalities | Mixed gonad dysgenesis (45X/46, XY) |
| Ovotesticular DSD | Ovarian and testicular tissue: many possible karyotypes |
Sex chromosome disorders of sexual development—X-chromosome anomalies
Turner syndrome
Turner syndrome results from the partial or complete absence of the short (“p”) arm of an X chromosome. The frequency of Turner syndrome is ~1 in 3000 live-born females. Approximately 5%–10% of recognized first-trimester spontaneous abortuses are affected with Turner syndrome.
Only about 1% of Turner conceptuses come to term.
About half of girls with Turner syndrome exhibit a 45, X karyotype. Other Turner karyotypes include: 46, X,i(Xq) (an isochromosome of the long arm of the X chromosome), 46, X, del(Xp) (a.k.a. 46, XXp-; an X-chromosome short-arm deletion), 46, X,r(X) (a ring chromosome where the telomeres of the short and long arms of the X chromosome are “fused”) and various mosaics (e.g., 45, X/46, XX; 45, X/47, XXX).
On a molecular basis, the most important cause of Turner syndrome is haploinsufficiency (one copy) of the short stature homeobox ( SHOX ) gene (chromosome Xp22.33). This gene is a member of the paired homeobox family of transcription factors, which is highly conserved across species (e.g., mammals, fish, and flies). SHOX is located in the pseudoautosomal region 1 (PAR1) of the X and Y chromosomes. SHOX regulates fundamental events in development and growth. Normal males and females have two functional copies of SHOX . SHOX mutations can be seen in cases of idiopathic growth retardation, Léri–Weill dyschondrosteosis (where 1 SHOX copy is deleted), and Langer mesomelic dysplasia (where both SHOX copies are deleted).
There is no genital ambiguity in individuals with Turner syndrome; however, the gonads do not form normally and the result is “streak” gonads (e.g., a type of aplasia where the ovary is replaced by functionless tissue with total or near exhaustion of oocytes). These gonads are greatly depleted of ova so that most girls with Turner syndrome do not estrogenize at the time of puberty (e.g., there is primary hypogonadism, a.k.a. there is a failure of “gonadarchy”). However, adrenal androgens are produced at the time of puberty resulting in axillary and pubic hair (e.g., there is evidence of normal “adrenarchy” that, together with “gonadarchy,” constitutes the two key processes of normal puberty). Overall, 5%–10% of girls with Turner syndrome do estrogenize at the time of puberty, and 2%–5% of girls with Turner syndrome do become pregnant ; however, there is a high risk of fetal loss, and congenital and chromosomal anomalies .
Besides primary hypogonadism, other anomalies can occur including short stature (~100% frequency), lack of a growth spurt at the time of puberty, perceptive hearing impairment (50% frequency), micrognathia (>70% frequency), anomalous ears (80% frequency), webbing of the neck, high arched palate, a low hairline at the back of the neck, increased frequency of Hashimoto thyroiditis, widely spaced nipples, a shield (broad) chest, bicuspid aortic valve, coarctation of the aorta, lentigines (freckle-like brown spots) on the trunk, horseshoe kidney, hyperconvex nails, short fourth or fifth metacarpals or metatarsals, and increased carry angle at the elbows. Cystic hygromas of the neck and swellings of the back of the hands or the top of the feet can be seen at birth, although most girls are phenotypically normal at birth. Girls with Turner syndrome display normal intelligence; however, some girls may be challenged by spatial concepts. Noonan syndrome is sometimes confused with Turner syndrome. Noonan syndrome affects males or females and is inherited as an autosomal dominant trait.
Laboratory findings –
At the time of puberty, estradiol is low and gonadotropins are elevated. The diagnosis is made via the karyotype.
Trisomy (triple) X
Affecting approximately 1 in 100 females, trisomy (triple) X results when there are three copies of the X chromosome (47, XXX). Stature can be increased in trisomy X; however, there are no typical phenotypic anomalies and there is no sexual ambiguity. Delayed development (e.g., delayed motor milestones, poor coordination, and awkwardness) and potentially serious learning problems (~70% frequency) can be present. Infertility can occur. More than half of affected individuals require special education in high school. Nevertheless, most affected individuals cope well and adapt as independent adults. Offspring are almost always chromosomally normal. An increasing number of X chromosomes [e.g., tetrasomy X syndrome (48, XXXX) or pentasomy X syndrome (49, XXXXX)] produce increasing severities of developmental impairment and physical problems.
Laboratory findings –
If hypogonadism is present, the estradiol is expected to be low with elevated gonadotropins (e.g., primary hypogonadism). The diagnosis is made via the karyotype. This condition could be identified during an evaluation for infertility . Some 47, XXX women have had hypogonadotropic hypogonadism . This is a result for gonadotropin-releasing hormone (GnRH) deficiency due to a coexisting chromosome 4 partial deletion.
46, XX sex-reversed male
A “46, XX sex-reversed male” is a term that applies to males who display a 46, XX karyotype. Sometimes the term “sex-reversal” is used in place of “sex-reversed.” In this chapter, such individuals are classified as having a sex chromosome DSD because the defect in the majority of cases is in the X chromosome.
The phenotype is usually male with small testes, laboratory evidence of mild hypogonadism (in pubertal and adult males: elevated gonadotropins and low testosterone), azoospermia, and infertility in adolescence/adulthood. A male phenotype is present because one of the X chromosomes has acquired Y-chromosome material during spermatogenesis in the father. Sufficient Y-chromosome material is present to foster the bipotential gonad developing into a rudimentary testis.
The Y-chromosome genes that aberrantly crossover to the X chromosome are near the pseudoautosomal region where normally the X and the Y chromosomes do crossover during meiosis. With sufficient Y-chromosome material on an X chromosome, a testis can form in utero causing virilization. Because of two X chromosomes, similar to a male with Klinefelter syndrome (47, XXY), the testicular tubules do not form properly causing reduced testicular size and infertility.
80%–90% of men with the 46, XX karyotype are SRY positive . Sex-reversed males who lack the SRY gene may or may not exhibit ambiguous genitalia .
Not due to a sex chromosome aberration, there is a report of an infant with severe penile/scrotal hypospadias whose mosaic karyotype was 46, XX, dup(17)(q23.1q24.3)/46, XX . SRY was absent. FISH demonstrated a duplication of SOX9 . The testes-promoting effect of multiple expressed copies of SOX9 might be predicted as SRY expression regulates SOX9 expression . This has also been reported in an Italian family . While these cases do not involve defects in a sex chromosome (and actually deserve placement in the section on 46, XX DSDs), it seemed most pragmatic to discuss these interesting cases with the majority of 46, XX sex-reversed male cases.
Laboratory findings –
Most commonly 46, XX males present with small testes and/or infertility and are SRY positive. Mild primary hypogonadism is detected. These men will be infertile.
Sex chromosome disorders of sexual development—Y-chromosome anomalies
Klinefelter syndrome
Affecting 1 in 1000 males, Klinefelter syndrome results from the 47, XXY karyotype where males have an extra X chromosome. This interferes with normal testicular development, structure, and function [producing gonadal (testosterone) deficiency], has adverse effects on intellectual development, and produces several phenotype anomalies.
Usually there is no severe sexual ambiguity; however, cryptorchidism, hypospadias, and micropenis may be observed. Puberty can be delayed from hypogonadism. The testes in adult men display fibrosis and hyalinization of the seminiferous tubules, are reduced in size and exhibit azoospermia (absence of sperm production) causing infertility.
Males affected with Klinefelter syndrome can exhibit behavioral problems including “dullness,” autism, psychosocial abnormalities (e.g., excessive shyness), language and learning disabilities, gynecomastia (from testosterone deficiency), an increased risk for breast cancer (although the degree of increased risk is not well defined ), tall stature (with long bone abnormalities), a decreased upper/lower segment ratio, arm span exceeding height by more than 2 inches, and an increased risk of Hashimoto thyroiditis and systemic lupus erythematosus. Many other complications can occur later in life (e.g., pulmonary diseases, cancers, varicose veins causing leg ulcers, and diabetes mellitus).
Laboratory findings –
With hypogonadism, testosterone is reduced with elevated gonadotropins (e.g., primary hypogonadism). The diagnosis is made via the karyotype.
47, XYY
The phenotype of men with the 47, XYY karyotype is usually normal (including fertility). 47, XYY is equal in frequency to Klinefelter syndrome. Thus the overall frequency of sex chromosome anomalies in men is approximately 1 in 500. Stature is usually increased in men with a 47, XYY karyotype. Sometimes there are adverse psychological occurrences (e.g., impulsivity, conduct/behavioral difficulties or attention deficits), delayed development of language and speech skills, learning disabilities, delayed motor skills, hypotonia, and various bone and skeletal anomalies (macrodontia, flat feet, clinodactyly, hypertelorism, and scoliosis).
Laboratory findings –
Gonadal function is normal. The diagnosis is made via the karyotype.
46, XYp-
Deletions of the short arm of the Y chromosome (46, XYp-) with loss of the key SRY gene leads to a female phenotype with failure to enter puberty . Not all such girls will manifest a Turner-syndrome-like phenotype . An isochromosome of the long arm (q) of the Y chromosome [46, X,i(Yq)] can cause a similar phenotype with two copies of the Y-chromosome long arm (q) and no copies of the Y-chromosome short arm (p).
“46, XYp–” can be viewed as a form of gonadal dysgenesis . However, because the aberration is in a sex chromosome, such individuals are classified in this chapter as having a “sex chromosome DSD – Y chromosome anomaly.”
Laboratory findings –
The diagnosis is dependent upon the karyotypic analysis. There will be pubertal failure, infertility, and hypergonadotropic hypogonadism.
46, XY sex-reversed female
A “46, XY sex-reversed female” is a term that applies to females who display a 46, XY karyotype when other causes of a DSD have been excluded. Similar to 46, XX sex-reversed males, in this chapter, 46, XY sex-reversed females are classified as having a Y-chromosome DSD because the defect in the majority of cases is in the Y chromosome.
This condition is karyotypically different from 46, XYp- because in a 46, XY sex-reversed female, the Y chromosome is normal by G-banding, yet there is a loss of genes or mutation in genes (such as SRY ) that normally determine the development of the testis . When Y-chromosome material is lost (presumably during gametogenesis) or there is an SRY loss-of-function mutation, the testes do not form normally resulting in a female phenotype. However, because the gonad is not a normal ovary, the affected individual will not enter into puberty . There should also be concern for a gonadal neoplasm since the intraabdominal gonad has a Y chromosome. Therefore gonadectomy should be performed.
46, XY sex-reversed females may also result from terminal deletions of distal chromosome 9p . In the rare condition, campomelic dysplasia, affected individuals with a 46, XY karyotype can be phenotypically female due to SOX9 mutations .
Laboratory findings –
Most commonly 46, XY sex-reversed females present with failure to enter puberty. SRY is mutated or absent. SRY gene sequencing is commercially available. Rarely defects in SOX9 are present, but in such cases, severe skeletal anomalies are recognized. Like so many other conditions, other causes of a 46, XY DSD must be considered and excluded.
Sex chromosome disorders of sexual development—X- and Y-chromosome anomalies
Mixed gonad dysgenesis
Mixed gonad dysgenesis is characterized by the mosaic karyotype of 45X/46, XY . Typically, there is a testis on one side and a streak gonad on the other side. When an individual’s two gonads are not the same, the term “asymmetrical gonadal dysgenesis” can be applied, although this is not specific to mixed gonadal dysgenesis as “asymmetrical gonadal dysgenesis” can occur in cases of ovotesticular DSD (see below).
Ambiguous genitalia is frequently observed in persons with mixed gonadal dysgenesis. An important concern is the risk of a gonadal neoplasm (e.g., germ cell tumor) developing in a person who has a “dysgenetic” gonad (an abnormal gonad with Y-chromosome material) .
Laboratory findings –
The diagnosis is made via the karyotype in an individual with masculinized yet ambiguous genitalia.
Sex chromosome disorders of sexual development—ovotesticular disorders of sexual development
Strictly speaking, ovotesticular DSD is a condition characterized the presence histologically of both ovarian (i.e., follicles) and testicular (i.e., tubules) tissue. Ovotesticular DSD, therefore, is defined by histology and not the karyotype. Ovotesticular DSD is different from “mixed gonadal dysgenesis” where one gonad is a testis and the other gonad is a streak. In ovotesticular DSD, various combinations of gonads can be found as summarized in Table 16.2 . Genital ambiguity can be present. Some authors treat ovotesticular DSD as a separate category of DSD in parallel with sex chromosome DSDs, 46, XY DSDs, and 46, X DSDs .
| Ovatestis | Ovatestis |
| Ovatestis | Absent |
| Ovatestis | Ovary |
| Ovatestis | Testis |
| Testis | Ovary |
Noting that it is the gonadal histology that defines an ovotesticular DSD and not the karyotype, a variety of karyotypes are observed in affected individuals such as 46XX (most common), 46XY (second most common), 46XX/46XY, and 45X/46XX. It is unclear to what degree the 46XX/46XY and 45X/46XX karyotypes represent mosaicism (derived from a single conceptus) versus chimerism (derived from more than one conceptus). Note that ovotesticular DSD will also come under the headings of “46, XY DSDs” and “46, XX DSDs”; however, this discussion of ovotesticular DSD will also serve those categories for simplicity.
The mechanism of concurrent testicular and ovarian development in ovotesticular DSD is known in some cases. Individuals with ovotesticular DSD with the 46, XX karyotype may (1) carry an SRY gene that has crossed over from a Y chromosome to an X chromosome, (2) display a SOX9 duplication yielding three copies , (3) exhibit “hidden” mosaicism (a Y-bearing cell line is present that is not detected by karyotype) , (4) include a homozygous missense mutation in the R-spondin1 Gene (RSPO1; chromosome 1p34.3) , or (5) encompass an NR5A1 mutation that encodes SF-1 .
Laboratory findings
The diagnosis is made via gonadal biopsy. Because gonadal biopsy is not routinely performed in cases of DSD, ovotesticular DSD would be considered (1) when other diagnoses have been excluded or (2) in cases where the gonads are asymmetric.
Ovotestes can fall into two major patterns: mixed type (admixed or compartmentalized; 90% of cases) or bipolar (10% of cases). Therefore the surgeon should biopsy each gonad from pole to pole so as not to miss an ovotestes. If the ovotestis is of the mixed type, such gonads should be removed. If polar in distribution, ovary-sparing surgery can be attempted.
Sex chromosome disorders of sexual development—summary
Cases of Turner syndrome, 47, XXX, 47, XXY, and 47, XYY do not usually display ambiguous genitalia and therefore present for other reasons, for example, webbing of neck, edema of the hands and feet, short stature and absent puberty in Turner syndrome; small testes, low testosterone or infertility in 47, XXY; and tall stature; learning disabilities; delayed speech, language, and motor skills; hypotonia; hand tremors/motor tics, and behavioral/emotional difficulties in 47, XYY. On the other hand, mixed gonadal dysgenesis (45X/46, XY) and ovotesticular DSD can present with ambiguous genitalia. While chromosomal studies provide the most diagnostic information (and define the disorder in cases of Turner syndrome, 47, XXX, 47, XXY, 47, XYY, and 45X/46, XY), evaluation of sex hormone concentrations and gonadotropins can indicate if primary hypogonadism is present. Table 16.3 summarizes these disorders.
| Condition | Karyotype | Ambiguous genitalia | Pubertal a | Laboratory findings | Comments | |
|---|---|---|---|---|---|---|
| Failure | Infertility a | |||||
| Ambiguous genitalia absent | ||||||
| Turner syndrome | 45, X, etc. | No (female) | Usually | Usually | HH | Distinct phenotype |
| Trisomy X | 47, XXX | No (female) | No | ± | ±HH | No distinct phenotype; HypoH possible |
| 46, XX sex-reversed male | 46, XX | Usually no (male) | No | Yes | HH | Small testes |
| Klinefelter syndrome | 47, XXY | No (male) | No | Yes | HH | Characteristic phenotype |
| 47, XYY | 47, XYY | No (male) | No | No | Nl | Tall, ±abn develop., and bone; ±cryptorchidism, hypospadias, or micropenis |
| 46, XYp- | 46, XYp- | No (female) | Yes | Yes | HH | ±Turner-like phenotype |
| 46, XY, sex-reversed female | 46, XY | No (female) | Yes | Yes | HH | No distinct phenotype |
| Ambiguous genitalia present | ||||||
| Mixed gonadal dysgenesis | 45X/46, XY | Common | – | – | Variable | 1 streak gonad, 1 testis |
| Ovotesticular DSD | Variable | Common | – | – | Variable | Dx requires gonad biopsy |
a When ambiguous genitalia are present, the patient should come to clinical recognition as a newborn. Pubertal failure and infertility are likely but the possibility of each depends upon the therapy.
46, XY disorders of sexual development
These DSDs result (1) from defects in the androgen–androgen receptor (AR) axis or (2) represent isolated and/or complex genitourinary malformations in 46, XY individuals. Testosterone synthesis, testosterone conversion to DHT and androgen binding to and activation of the AR constitute the androgen–AR axis. Inadequate androgen action can cause incomplete virilization of the genitalia in utero causing genital ambiguity. Table 16.4 classifies these defects.
Androgen deficiency
|
46, XY disorders of sexual development—failure of normal testicular development
There are four major conditions causing failure of testicular development. These include the following:
- •
complete gonadal dysgenesis;
- •
partial gonadal dysgenesis (some testicular tissue is present);
- •
gonadal regression; and
- •
ovotesticular DSD (discussed previously).
Complete gonadal dysgenesis
In complete gonadal dysgenesis (also known as Swyer syndrome), the testes never form. Female external and internal genitalia are present despite the 46, XY karyotype (e.g., there is no sexual ambiguity). Both testosterone and AMH are absent in normal concentrations in utero . This disorder most commonly comes to clinical recognition during the evaluation of girls who fail to enter puberty. Because of the presence of a Y chromosome, gonadectomy should be performed to prevent gonadal neoplasms (note: a detailed discussion of when gonadectomy should be performed is beyond the scope of this chapter). Because the uterus is structurally normal in complete gonadal dysgenesis, assisted reproduction through oocyte donation is an option for affect patients who desire pregnancy. The term “sex-reversed female” is sometime used to describe women with a 46, XY karyotype and complete gonadal dysgenesis. However, in complete gonadal dysgenesis, the Y chromosome is phenotypically normal.
Complete gonadal dysgenesis can be caused by loss-of-function mutations in SRY , NR5A1 (encoding SF-1), or DHH (desert hedgehog signaling molecule; chromosome 12q13.12), duplication of WNT4 (Wnt family member 4; chromosome 1p36.12) or DAX1 (a.k.a. NR0B1 gene; nuclear receptor subfamily 0 group B member 1, which antagonizes SRY ; chromosome Xp21.2), or gain-of-function mutations in MAP3K1 (mitogen-activated protein kinase kinase kinase 1; chromosome 5q11.2) .
Laboratory findings –
There is laboratory evidence of primary hypogonadism at the time of puberty (low testosterone compared to males and low estradiol compared to females; elevated LH and FSH). The karyotype is 46, XY. By FISH, the SRY gene may or may not be detected. Commercial SRY, NR5A1 , and DHH gene sequencing is available .
Partial gonadal dysgenesis
In partial gonadal dysgenesis, some testicular tissue is present. Because of the production of testosterone at reduced concentrations compared to normal in utero concentrations, ambiguous genitalia can result from inadequate virilization of the genitalia and inadequate AMH. Partial gonadal dysgenesis can result from SRY mutations, DAX1 duplications on the X chromosome, loss-of-function mutations in NR5A1 (SF-1; which can also cause adrenal insufficiency), or WT1 mutations (a tumor suppressor transcription factor that explains some cases of Wilm tumor; chromosome 11p13) .
Laboratory findings –
There is laboratory evidence of primary hypogonadism (low testosterone for a male; elevated LH and FSH) in a patient with ambiguous genitalia. The karyotype is 46, XY. Commercial SRY , NR5A1 , and WT1 gene sequencing is available .
Gonadal regression
Gonadal regression (the “vanishing” testes syndrome) is the condition where a male has fully virilized in utero but functioning testes are absent at the time of birth . During the evaluation of infants or boys with bilaterally undescended testes (bilateral cryptorchidism), the failure to identify normally formed testes leads to this diagnosis. The histological gonadal finding is typically a fibrovascular nodule with evidence of dystrophic calcification and hemosiderin-laden macrophages. Without exogenous testosterone administration at the time of puberty, there will be pubertal failure. A discussion of the structure of the testes in sporadic cryptorchidism is beyond the scope of this review. It is important to note that there are no reported occurrences of gonadal malignancies in gonadal regression cases suggesting that surgical gonadal removal may not be required.
Disappearance of the testes is ascribed to some sort of vascular “accident” in utero . There are no known biochemical or genetic causes for gonadal regression. If the testes “regress” during to or prior to development of the genitalia, ambiguous genitalia or a female phenotype can result . This is reminiscent of Swyer syndrome when an individual has a 46, XY karyotype, appears female, and lacks gonads.
Laboratory findings
We are now at the point in our discussions of DSDs where we tackle the question: “ Is functional testicular tissue present in an individual ?” This is relevant for many types of DSDs including gonadal regression. It must also be stated that the presence of testicular tissue does not mean than ovarian tissue is absent nor that ovotestes are absent. The evaluation of nontesticular gonadal tissue would require biopsy and histologic review.
Imaging (e.g., MRI) plus biopsy (either an open surgical biopsy or a needle biopsy) of the gonads (or gonadal remnants) can reveal their anatomy and histology but imaging does not reveal gonadal function. Therefore laboratory studies are required to determine if functional testicular tissue is present.
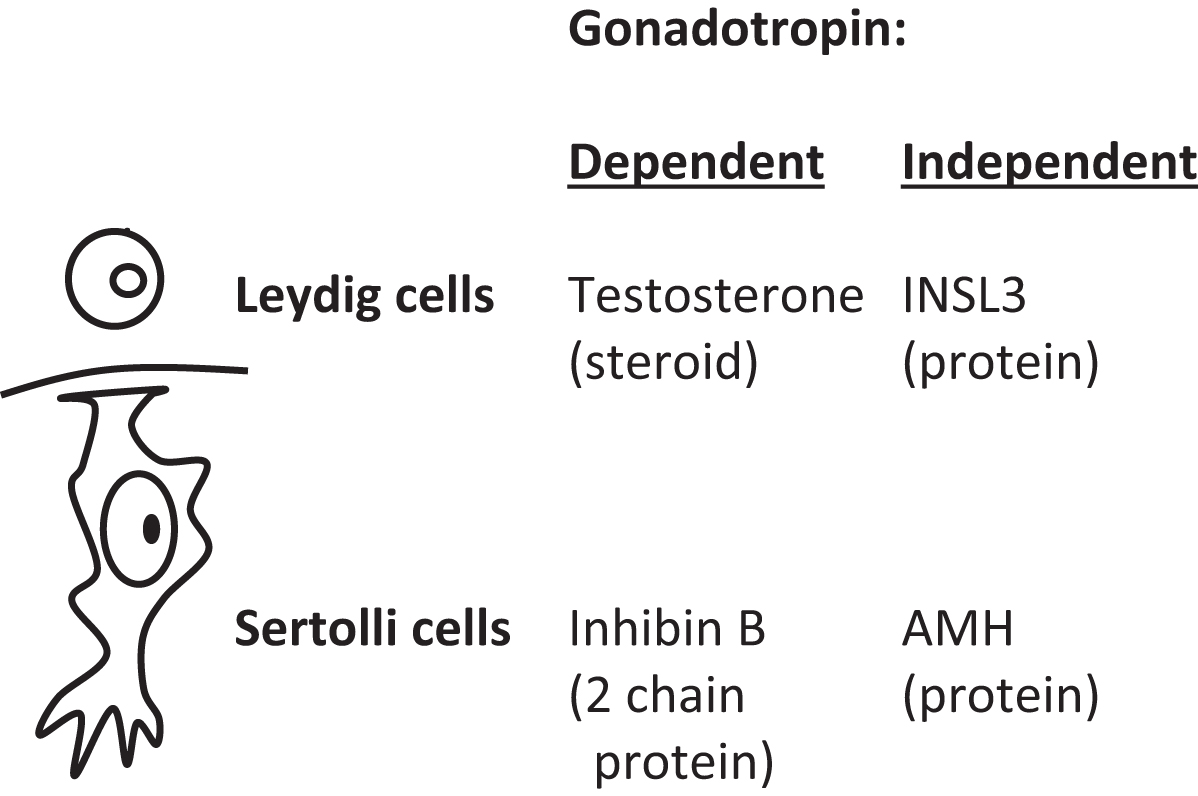
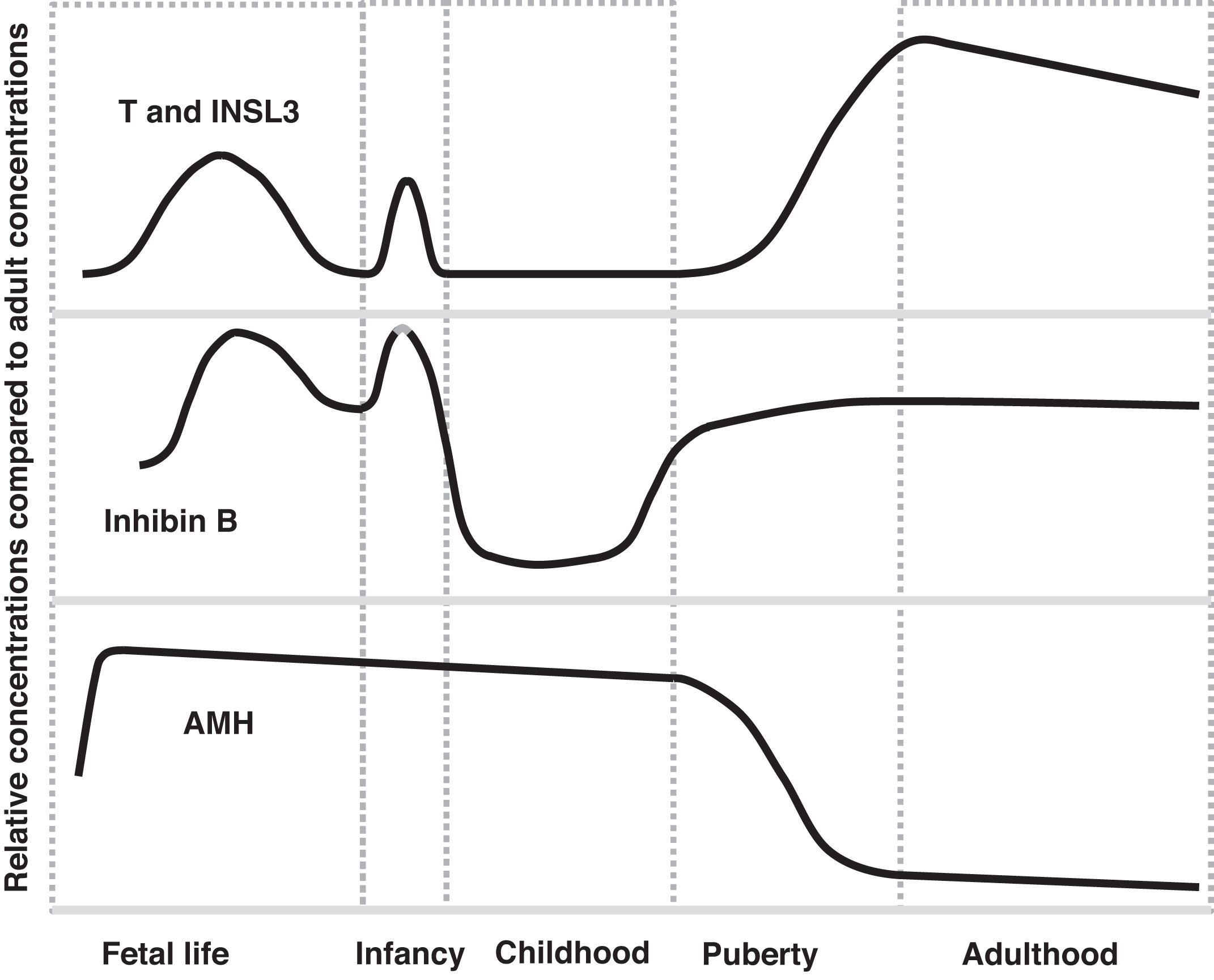
Plasma markers of Leydig cell function are testosterone and insulin-like 3 (INSL3; 131 amino acids). The gene for INSL3 is INSL3 located on chromosome 19p13.11. Markers of Sertoli cell function are inhibin B and AMH ( Fig. 16.4 ). Inhibin B is composed of two chains: an α chain of 366 amino acids that is shared with inhibin A [encoded by INHA (inhibin subunit alpha) gene (chromosome 2q35)] and a unique β B chain of 407 amino acids [encoded by inhibin subunit Beta B ( INHBB ) gene (chromosome 2q14.2)]. Testosterone and inhibin B are regulated through the hypothalamic-pituitary gonadotropin axis (a.k.a. “gonadotropin dependent”); in contrast, INSL3 and AMH are not similarly regulated (“gonadotropin independent”). Unique patterns of secretion between fetal life and adult life are displayed by many of these markers ( Fig. 16.5 ).
Testosterone—a Leydig cell marker
Testosterone’s and DHT’s effects on the development of the genitalia were described above. During later gestation, testosterone declines in males because placental estrogens suppress fetal gonadotropin (LH and FSH) secretion. In normal male infants, following delivery, the withdrawal of the gonadotropin-suppressive effects of in utero placental estrogens allows an immediate rise in LH for ~12 h (with a rise in testosterone) . Thereafter rising concentrations of LH and FSH continue that peak at 4–10 weeks then decrease to prepubertal levels by 6 months. This later decline in LH and FSH is ascribed to increasing sensitivity of the hypothalamus to sex steroid negative feedback .
As a result of these changes in gonadotropins following birth, testosterone peaks at ~3 months of age reaching concentrations observed in mid-pubertal males. By 6–9 months, testosterone concentrations decline to expected prepubertal concentrations. This transient rise in testosterone during infancy is termed “mini puberty” . The testosterone elevations of mini puberty may contribute to masculinization of the brain.
The importance of this physiology is that searching for biochemical evidence of a testis in an infant under 6–9 months of age (especially between 4- and 8-week age) may only require a testosterone measurement (note: while immunoassays may be satisfactory, if the testosterone levels are low, mass spectrometry would be required). However, a low “random” testosterone concentration does not absolutely exclude the presence of testicular tissue. Therefore, if the testosterone concentration is low, exogenous hCG can be administered and the testosterone subsequently measured poststimulation in search of evidence of testicular tissue. In such cases, hCG has an action similar to LH stimulating the Leydig cells to produce testosterone. Alternatively, another measure of testicular tissue can be sought (e.g., INSL3, AMH, or inhibin B). AMH and inhibin B assays are commercially available. The authors are not aware of FDA-approved methods to measure INSL3.
There are several protocols available where hCG is injected and some days later, blood for testosterone measurement is taken. Some authors define a response indicating the presence of testicular tissue as an increment in testosterone of 260 ng/dL or more . Another publication stated that a normal response to hCG injections in teenage boys was a rise in testosterone to greater than 200 ng/dL .
In terms of an hCG protocol, Grant et al. injected 1000 U of hCG daily and after 3 days identified significant testosterone increases. In boys with cryptorchidism, testosterone concentrations rose into the adult male range with either of two protocols: 1500 IU every other day for seven injections or 100 IU per kilogram every 4 or 5 days for a total of four injections . Adiyaman et al. compared two protocols. Using 3000 IU of intramuscular (IM) hCG per meter squared daily for 3 days, testosterone peaked 4 days after the last injection. Using 1500 IU of IM hCG per meter squared weekly for 3 weeks, testosterone peaked 1 day after the last injection. Tamunopriye and Abiola administer 100 IU of hCG per kilogram and measure testosterone 72–120 h later . A protocol from Leeds General Infirmary and Harrogate General Hospital recommends, in infants, 1500 U of hCG or, in boys over age 2 years, 5000 U of hCG is injected subcutaneously or intramuscularly followed by androgen measurements (testosterone, androstenedione, and DHT) on day 5.
Use of testosterone measurements as a testicular marker –
Measuring testosterone is always important in the evaluation of male hypogonadism and in many cases of DSD. If testosterone is deficient or absent, (1) a problem with gonadal development, (2) a problem with gonadal stimulation by gonadotropins, or (3) a problem with testosterone biosynthesis is present.
INSL3—a Leydig cell marker
A member of the insulin-like hormone superfamily, INSL3 is secreted exclusively by Leydig cells. INSL3 and its receptor relaxin family peptide receptor 2 (RXFP2; RXFP2 gene: chromosome 13q13.1) may play a role in normal testicular descent into the scrotum in utero , although this is controversial .
The INSL3 concentration rises in early infancy similar to testosterone (see mini puberty discussion above), regresses to low concentrations thereafter until late childhood, and then rises throughout puberty to adult levels. Therefore changes in INSL3 levels parallel changes in testosterone concentrations throughout life.
Because INSL3 is expressed and secreted constitutively, INSL3 levels are not regulated by the hypothalamic-pituitary-gonadotropin axis similar to the regulation of testosterone . Low levels of INSL3 during childhood are explained by the fact that the testis in childhood is predominantly composed of tubules with a lower proportion of Leydig cells. Regarding females, a role for INSL3 in antral follicle (a.k.a. Graafian or tertiary follicle) development is under investigation .
Use of INSL3 as a testicular marker –
Because INSL3 is not subject to control via LH, and INSL3 concentrations are relatively stable, theoretically INSL3 concentrations could be a superior marker for the presence or absence of the testes . This being said, INSL3 measurements are not available from major reference laboratories limiting their application in current day clinical practice.
Inhibin B—a Sertoli cell marker
Inhibin B is composed of two chains. The alpha chain is shared with inhibin A. The beta chain is unique (β B ). Both inhibin B and testosterone are controlled by the hypothalamic-pituitary-gonadotropin axis. Whereas testosterone (from Leydig cells) negatively feedbacks on the secretion of LH, inhibin B (from Sertoli cells) negatively feedbacks on the secretion of FSH.
Similar to the rise in testosterone with mini puberty, inhibin B rises transiently early in life. Inhibin B concentrations are high for the first 2 years of life, decline thereafter during childhood, and then rise during puberty to adult concentrations.
Normal or near-normal concentrations are observed in boys with cryptorchidism. Inhibin B is being investigated as a predictor of testicular function after orchiopexy .
Use of inhibin B as a testicular marker –
In the absence of testicular tissue, undetectable or low levels of inhibin B are recognized. However, if the Sertoli cells are normally present, even in the absence of Leydig cells, inhibin B levels should be normal. Because inhibin B is controlled by FSH, hypogonadotropic hypogonadism will result in low inhibin B. This emphasizes the point that gonadotropin measurements should accompany sex steroid measurements.
Anti-Mullerian hormone—a Sertoli cell marker
As discussed above, AMH is a product of Sertoli cells. AMH’s function in utero is to suppress the development of Mullerian structures. AMH rises in fetal life with the development of the testes (similar to rising testosterone levels in males). The concentrations fall somewhat after birth, and then decline to some degree throughout infancy and childhood and then decline further during puberty to low concentrations throughout adulthood. During puberty, AMH is suppressed by testosterone . This explains declining AMH during puberty as testosterone rises.
Use of anti-Mullerian hormone as a testicular marker –
The strength of AMH as a testicular marker is that AMH is relatively high during childhood in boys whereas testosterone, INSL3, and inhibin B levels in childhood are all relatively low. However during puberty, AMH is not a good marker of Sertoli cells as AMH levels decline during puberty. Therefore testosterone, INSL3, and inhibin B can be considered to constitute one set of postinfancy markers that are low until puberty and rise during puberty whereas AMH is high postinfancy until puberty.
46, XY disorders of sexual development—gonadotropin deficiency
If gonadotropins are deficient, testosterone may be produced in inadequate concentrations to fully virilize the developing 46, XY embryo . In newborn 46, XY individuals with ambiguous genitalia, evidence of craniofacial midline defects can suggest hypopituitarism and hypogonadotropic hypogonadism. Such midline defects can include cleft lip and/or palate, pituitary aplasia or hypoplasia, septo-optic dysplasia, holoprosencephy, and even anencephaly . Defects in a variety of genes including PIT1 (POUF1 ), PROP1 , LHX3 , LHX4 , and HESX1 have been reported to cause anterior pituitary hormone defects. Isolated gonadotropin deficiency can result from mutations in KAL and KISS1R . Further discussion of causes of gonadotropin deficiency is beyond the scope of this chapter on DSDs.
Laboratory findings
A discussion on hypopituitarism can be found in Chapter 2 .
46, XY disorders of sexual development—inborn errors in testosterone synthesis
Leydig cells synthesize testosterone from cholesterol ( Fig. 16.6 ). CYP17, 17-ketosteroid reductase, and 3 beta-hydroxysteroid dehydrogenase-delta 4,5 isomerase catalyze this conversion. CYP17 ( CYP17A1 , cytochrome P450 family 17 subfamily A member 1; chromosome 10q24.32) has two activities: 17-hydroxylase and 17,20-desmolase activities. All of the steroid synthesizing genes are autosomal and thus defects involving these genes are inherited most commonly as autosomal recessive traits.