Abstract
Disorders of sex development (DSD) are rare disorders occurring when there is a discordance between chromosomal, gonadal, or phenotypic sex. These occur in the presence of genetic mutations that affect one of the two major processes in sex development: sex determination or sex differentiation. In sex determination, the bipotential gonad is genetically programmed based on the sex chromosome complement to become either a testis or ovary. Sex differentiation occurs in the presence of a formed testis or ovary, and is dependent upon the ability of the gonad to produce hormonal factors and/or the presence of the appropriate receptors in extragonadal tissues.
Keywords
disorders of sex development, testis, ovary, sex determination, sex differentiation, sex chromosome, ambiguous genitalia
Disorders of sex development
“Is it a boy or a girl?” is probably the most frequently asked question to a new mother. To this apparently simple question, the answers can become rather complicated. First, one could try to define precisely what “boy or girl” entails. In other words, how do we define sex? Although common sense would dictate that the appearance of the external genitalia should define what sex really is, biological complexity suggests otherwise. Many biological parameters are crucial to precisely delineate the sex of an individual: chromosomal constitution (46,XX or 46,XY), sex determining genes (presence or absence of Sry), gonadal histology (testis or ovary), hormonal output (testosterone or estradiol), sex of internal reproductive organs (uterus, fallopian tubes, or prostate, epipidymis, vas deferens). Each of these parameters can be disrupted in disorders of sex development (DSD), defined as “congenital conditions in which development of chromosomal, gonadal or anatomical sex is atypical.”
DSD are typically categorized in disorders of sex determination and disorders of sex differentiation (see Fig. 19.1 ). In the former, the development of the gonads is disrupted, and leads to either gonadal dysgenesis (GD), or the development of ovotestis. In the latter, the development of the gonads is normal, but the fetus develops either more virilized than typical for a 46,XX individual (in the case of congenital adrenal hyperplasia (CAH) for instance, see below), or more feminized than a typical 46,XY individual (e.g., in the case of androgen insensitivity syndrome (AIS)). The goal of this chapter is to help the physician navigate the complexities of atypical sex development and identify rapid tools to diagnose patients with DSD.
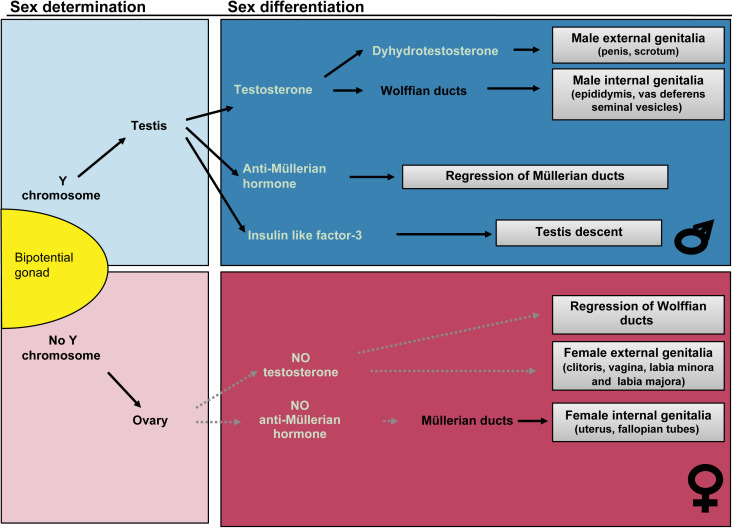
Disorders of sex determination
46,XY Disorders
Gonadal Dysgenesis
In 46,XY GD, testes fail to undergo normal development. 46,XY GD can be isolated or syndromic (see Table 19.1 ), and can be pure, partial, or mixed ( Table 19.2 ).
| Syndromes associated with ambiguous genitalia | Phenotype | Gene | Locus | OMIM # | |
|---|---|---|---|---|---|
| Syndromes associated with GD | Gonadal dysgenesis with adrenal hypoplasia | XY GD with adrenal hypoplasia. Gonadal dysgenesis due to SF1 mutations can also be isolated | SF1/NR5A1 | 9q33 | 184757 |
| Denys–Drash | XY GD, early diffuse mesangial sclerosis of kidneys, Wilms’ tumor | WT-1 | 11p13 | 194080 | |
| Frasier | XY GD, early adolescent development of focal segmental glomerulosclerosis, gonadoblastoma | WT-1 | 11p13 | 136680 | |
| Campomelic dysplasia | Ambiguous genitalia, congenital bowing of long bones, hypoplastic scapulae, and thoracic vertebrae pedicles | SOX9 | 17q24.3-q25.1 | 114290 | |
| GD with neuropathy | XY GD with associated minifascicular neuropathy (one of four cases) | DHH | 12q12-13.1 | 607080 | |
| X-linked alpha-thalassemia/mental retardation | Hemoglobin H disease, mental retardation, dysmorphic facies, genital abnormalities | XH2 | Xq13.3 | 301040 | |
| Palmoplantar hyperkeratosis with squamous cell carcinoma and XX sex reversal | Variable degrees of XX sex reversal, palmoplantar keratoderma, squamous cell carcinoma, congenital bilateral corneal opacities, onychodystrophy, and hearing impairment | RSPO1 | 1p34.3 | 610644 | |
| Blepharophimosis-ptosis-epicanthus inversus syndrome type I | Blepharophimosis, ptosis, and epicanthus inversus syndrome, either with or without POF | FOXL2 | 3q23 | 110100 | |
| Syndromes associated with small penis and/or cryptorchidism | VACTERL/VATER | Vertebral defects, anal atresia, tracheoesophageal fistula with esophageal atresia, and radial dysplasia, limb anomalies | Unknown | Unknown | 192350 |
| Goldenhar syndrome (hemifacial microsomia) | Unilateral deformity of the external ear and small ipsilateral half of the face with epibulbar dermoid and vertebral anomalies | Unknown | 14q32 | 164210 | |
| Smith–Lemli–Opitz syndrome (SLOS) | Multiple congenital malformation and mental retardation syndrome | DHCR7 | 11q12-q13 | 270400 | |
| Pallister–Hall syndrome (PHS) | Hypothalamic hamartoblastoma, postaxial polydactyly, and imperforate anus | GLI3 | 7p13 | 146510 | |
| Robinow syndrome | Mesomelic limb shortening associated with facial and genital abnormalities | ROR2 | 9q22 | 180700 | |
| Prader–Willi syndrome | Obesity, muscular hypotonia, mental retardation, short stature, hypogonadotropic hypogonadism | SNRPN | 15q12, 15q11-q13 | 17620 | |
| Kallmann syndrome | Hypogonadotropic hypogonadism and anosmia | FGFR-1 | 8p11.2-p11.1 | 147950 | |
| Holoprosencephaly | Craniofacial dysmorphology | Many | 21q22.3, 2q37.1-q37.3 | 236100 | |
| Malpeuch facial clefting syndrome | Short stature, hypertelorism, eye anomalies, facial clefting, hearing loss, urogenital abnormalities, mental retardation | Unknown | Unknown | 248340 | |
| Najjar syndrome | Genital anomaly, mental retardation, and cardiomyopathy | Unknown | Unknown | 212120 | |
| Varadi–Papp syndrome | Big toes, hexadactyly, cleft lip/palate or lingual nodule, and somatic and psychomotor retardation. Some showed absent olfactory bulbs and tracts, cryptorchidism | Unknown | Unknown | 277170 | |
| Juberg–Marsidi syndrome | Cleft lip/palate with abnormal thumbs and microcephaly | Unknown | Unknown | 216100 | |
| Johanson–Blizzard syndrome | Aplasia or hypoplasia of the nasal alae, congenital deafness, hypothyroidism, postnatal growth retardation, malabsorption, mental retardation, midline ectodermal scalp defects, and absent permanent teeth | Unknown | 15q15-q21.1 | 243800 | |
| Borjeson–Forssman–Lehmann syndrome | Severe mental defect, epilepsy, hypogonadism, hypometabolism, marked obesity | Unknown | Xq26.3 | 301900 | |
| Torticollis, keloids, cryptorchidism, renal dysplasia | Torticollis, keloids, cryptorchidism, renal dysplasia | Unknown | Xq28 | 314300 | |
| Hypertelorism with esophageal abnormality and hypospadias | Laryngotracheoesophageal cleft; clefts of lip, palate, and uvula; swallowing difficulty and hoarse cry; genitourinary defects, mental retardation; congenital heart defects | Unknown | 22q11.2 | 145410 | |
| Faciogenitopopliteal syndrome | Cleft palate and webbing intercrural pterygium | Unknown | 1q32-q41 | 119500 | |
| Dubowitz syndrome | Short stature, microcephaly, mild mental retardation with behavior problems, eczema, and distinctive facies | Unknown | Unknown | 223370 | |
| Noonan syndrome | Hypertelorism, a downward eye slant, and low-set posteriorly rotated ears, short stature, webbed neck, cardiac anomalies | PTPN11 | 12q24.1 | 163950 | |
| Aarskog syndrome (faciogenital dysplasia) | Embryonic ocular hypertelorism, anteverted nostrils, broad upper lip, and peculiar penoscrotal relation | FGD1 | Xp11.21 | 305400 | |
| Cornelia de Lange syndrome | Low anterior hairline, anteverted nares, maxillary prognathism, long philtrum, “carp” mouth) in association with prenatal and postnatal growth retardation, mental retardation | NIPBL | 5p13.1 | 122470 | |
| Rubinstein–Taybi syndrome | Mental retardation, broad thumbs and toes, and facial abnormalities | CREBBP | 16p13.3, 22q13 | 180849 | |
| Seckel syndrome | Growth retardation, microcephaly with mental retardation, and a characteristic “bird-headed” facial appearance | SCKL1 | 3q22-q24 | 210600 | |
| Miller–Dieker syndrome | Microcephaly and a thickened cortex with 4/6 layers | LIS1 | 17p13.3 | 247200 | |
| Lenz–Majewski hyperostosis syndrome | High palate, short, yellow, carious teeth, progeroid appearance, short, increased venous pattern of the forehead and thorax | Unknown | Unknown | 151050 | |
| Lowe syndrome | Ophthalmic, cataract, mental retardation, vitamin D-resistant rickets, amino aciduria | OCRL1 | Xq26.1 | 309000 | |
| Syndromes associated with Müllerian malformation | MURCs | Müllerian duct aplasia, renal aplasia, and cervicothoracic somite dysplasia | Unknown | Unknown | 601076 |
| Mayer–Rokitansky–Kuster–Hauser syndrome (MRKH) | Müllerian duct aplasia | WNT4 in subset with hyperandrogenism | Unknown; 1p35 | 277000 | |
| McKusick–Kaufman syndrome (MKKS) | Hydrometrocolpos, congenital heart malformations, postaxial polydactyly | Unknown | 20p12 | 236700 |
| DSD | Biochemical changes | Differentiating features | Genetic diagnostic criteria | |
|---|---|---|---|---|
| DISORDERS OF SEX DETERMINATION | ||||
| 46,XY disorders | 46,XY pure/partial/mixed GD | Pure/partial/mixed: (+) FSH, LH Nml to (–) AMH No (+) with hCG Pure: (– –) T, DHT, E2 (– –) AMH Partial/Mixed: (–) T, DHT, E2 | Presence of partial testicular function (T, AMH) points towards partial or mixed GD. However, histological examination of testes after prophylactic gonadectomy can differentiate between pure, partial, and mixed GD. There is little genotype-phenotype correlation, but the presence of mosaicism 45,X/46,XY is often associated with mixed GD | Sequencing for SF1 and SRY mutations; if no mutation is found, CGH should be performed to look for copy number variants as a source for XY GD. If partial/mixed GD suspected, patient should be tested for mosaicism |
| 46,XX disorders | XX testicular/ovotesticular DSD | (+) FSH, LH (–) T, DHT Nml AMH No (+) with hCG | Testicular and ovotesticular DSD are at different ends of a phenotypic spectrum. The only way to definitively differentiate between 46,XX testicular and ovotesticular DSD is through complete gonadal histological examination looking for the presence of ovarian tissue | Testicular DSD: FISH for presence of SRY or SOX9. If clinical phenotype appropriate, sequence RSPO1 gene. If no molecular diagnostic is successful, CGH should be performed. Ovotesticular DSD: Search for XX/XY mosaicism and sequencing for SRY mutation |
| XX GD | (+) FSH, LH No (+) with hCG No AMH | Full female phenotype with amenorrhea and lack of secondary sex development | For cases of isolated 46,XX GD, sequencing for FSH mutations can be performed. If clinical presentation matches, sequence FOXL2 mutation. CGH can also be done to identify causative duplications or mutations | |
| DISORDERS OF SEX DIFFERENTIATION | ||||
| Disorders of androgen synthesis and action | StAR deficiency ( StAR) ; P450scc deficiency ( CYP11A1 ) | (+) Renin (–) Aldo (+) K (–) Na (–) All adrenal hormones (–) 17OH-P | Presence of lipid vacuoles in adrenals on histology. P450scc deficiency does present with enlarged adrenals. No HTN and hyperkalemia differentiates from CYP 17 deficiency | Presence of lipid filled vacuoles on histology; sequencing of the StaR or CYP11A1 genes and presence of a mutation in either one gives definitive diagnosis |
| 3βHSD type II ( HSD3B2 ) | (+) Renin; (–) Aldo, F (+) K, (–) Na (+) Ratio Δ 5 -17-pregnenolone: F (–) A, T | Baseline and ACTH-stimulated ratios of Δ 5 -17-pregnenolone:cortisol consistently distinguished between patients affected and nonaffected patients | Sequencing of the HSD3B2 gene | |
| 17α-Hydroxylase/17,20-lyase ( CYP17 ) | (–) Renin (–) Aldo, F (–)17OH-P (+) LH, FSH (+) Progesterone, DOC, B (+) Na (–) K (–) DHEA-S, A, T; No response to hCG stim | Hypertension and hypokalemic alkalosis in the presence of low 17-OH progesterone | Sequencing of the CYP17 gene | |
| POR | (+) 17OH-P (+) Progesterone (–) F, DHEA-S (–) A, T | Hypertension in the presence of elevated 17OH-P. Occasionally presence of Antley–Bixler skeletal malformations | Sequencing of CYP450 oxidoreductase | |
| 17βHSD type 3 ( HSD17B3 ) | Nml to ↑ A (+) Ratio A/T (>15) (–) T, DHT | Differentiate from 5α-reductase type 2 by levels and ratios of serum androgens | Sequencing of the HSD17B3 gene for deletions, insertions, and point mutations | |
| LCH | (+) LH Nml FSH (+) AMH (–) T, DHT, E 2 (–) hCG response Nml A/T ratio | To differentiate LCH from GD, AMH is used as a marker of testicular function | Sequencing of the LHCHR gene for deletions, insertions, and point mutations | |
| 5α-Reductase type 2 | Nml FSH, LH Nml T, E 2 ; (–) DHT (+) Ratio T/DHT (>30) | Development of male secondary sex characteristics in puberty with fine and sparse facial hair. Unlike HSD17B3 and AIS, no gynecomastia during puberty | Sequencing of the SRD5A2 gene | |
| AR | Nml FSH, LH (PAIS) (–) FSH, LH (CAIS) Nml to (+) AMH Nml A, T, DHT (+ +) hCG response | Female phenotype with breast development at puberty, with sparse pubic and axilla hair | Sequencing of AR gene looking for single amino acid substitutions, which account for 90% of reported cases | |
| AMH/AMHR | Nml hormonal profile | Presence of both Müllerian and Wolffian derivatives usually discovered incidentally | ||
| Disorders of androgen excess | 21α-Hydroxylase ( CYP21 ) | (+) Renin (–) Aldo, DOC, F (+) K; (–) Na (+) 17OH-P (+) DHEA-S, A, T | 17-OH progesterone elevated in the absence of hypertension, which is typical in HSD11B1 deficiency | Sequencing of CYP21 gene gives definitive diagnosis and can inform parents about genetic counseling in future pregnancies |
| 11βHSD1 ( HSD11B1) | (–) Aldo, F, Renin (+) DOC (+) K, (–) Na (+) 17OH-P (+) DHEA-S, A, T | Differentiate from CYP21 deficiency by presences of hypertension and hypokalemic alkalosis | Sequencing of the HSD11B1 gene | |
| P450 aromatase ( CYP19 ) | (+) 16OH-A (maternal) (+) FSH, LH, A, T (–) Estrone, E 2 | The presence of maternal virilization during pregnancy and XX virilization, which stops after delivery | Sequencing of CYP19 gives definitive diagnosis | |
Pure 46,XY GD is characterized by intra-abdominal bilateral fibrous streak gonads that fail to produce anti-Müllerian hormone (AMH) and testosterone, resulting in an unambiguous female phenotype (formerly Swyer syndrome), with an occasionally hypoplastic but typically well-formed uterus and fallopian tubes, and female external genitalia. Partial 46,XY GD entails varying amounts of testicular dysgenesis and ambiguous genitalia. Approximately 15% of pure and partial 46,XY GD is attributable to mutations in SRY , a transcription factor that targets another male-determining gene, SOX9. Over 50 mutations of the SRY open reading frame (ORF) have been identified in 46,XY GD, and deletions of Yp, a region of the Y chromosome containing SRY, have also been implicated. Most known mutations in SRY disrupt the high mobility group (HMG) box, which results in reduced nuclear import of SRY protein or impaired binding or bending of target gene DNA by SRY. Another 15% of pure and partial 46,XY GD is due to mutations in SF1 (NR5A1), an orphan nuclear receptor required for testis and adrenal development. SF-1 interacts with transcription factors GATA-4 and FOG-2 to regulate SRY expression in developing testes. Human mutations in GATA4 and FOG2 /ZFPM2 have recently been described in rare cases of 46,XY DSD. Most recently, MAP3K1 has been identified in a subset of patients with isolated 46,XY GD and knockdown of other Map-kinase factor mouse models have been shown to act upstream of Sry to promote its upregulation (Warr, 2012 ; Bogani, 2009 ). 46,XY GD due to SF1 haploinsufficiency can be isolated or syndromic ( Table 19.1 ). Very rarely, pure and partial 46,XY GD result from duplications of putative “antitestis” genes ( Rspondin1 , WNT4, DAX1 ), or mutations in genes necessary for testes organogenesis ( XH2, SOX9, WT1, DHH ) ( Table 19.1 ). Together, these genetic factors account for only about 40% of pure and partial 46,XY GD, indicating that additional, as yet unknown, genes are required for human testis determination.
Mixed 46,XY GD refers to asymmetric gonadal development resulting in asymmetric virilization of the external and/or internal genitalia and unilateral cryptorchidism. One gonad may be an abdominally located, fully dysgenic streak with no ipsilateral virilization. The contralateral testis appears normal to partially dysgenic, and its level of activity determines its location and the extent of virilization on that side. Typically, the etiology of mixed 46,XY GD is 45,X/46,XY mosaicism, causing a range of asymmetric patterns of male-determining gene expression and consequent phenotype. The proportion of Y chromosomal material in the gonad appears to correlate with the amount of testicular tissue and of phenotypic maleness.
Presentation and Diagnosis
Pure 46,XY GD should be considered when an adolescent presents as a phenotypic female with delayed puberty and primary amenorrhea. Pure 46,XY GD patients may be of normal to tall stature and have normal to small Müllerian structures, bilateral streak gonads, and no Turner stigmata. Rarely, they may present with a detectable abdominal or pelvic gonadoblastomal mass. Patients with partial or mixed 46,XY GD typically present much earlier than those with pure 46,XY GD – as infants or in early childhood – with ambiguity of internal and/or external genitalia. Patients with 45,X/46/46,XY mosaicism, in particular, can present with a wide range of phenotypes.
When the presumptive diagnosis suggests GD, and there are no indications of syndromic involvement ( Table 19.1 ), further criteria may strengthen the diagnosis of isolated 46,XY GD ( Table 19.2 and Fig. 19.2 a). The karyotype of peripheral leukocytes will show 46,XY for pure and partial GD, and, frequently, 45,X/46,XY mosaicism for mixed GD. Pelvic ultrasound may show bilateral atrophic gonads with a normal to hypoplastic uterus in the case of pure to partial 46,XY GD, but a unilateral streak gonad with asymmetric Müllerian and Wolffian structures in mixed GD. Diagnostic biochemical criteria are listed in Table 19.2 , and include elevated follicle stimulating hormone (FSH) and lutenizing hormone (LH). Fluorescent in situ hybridization (FISH) can be used to detect SRY or Yp, and sequencing the ORF is typically performed in all patients with 46,XY GD. However, given the advent of exome sequencing for clinical genetic diagnosis and the ever-increasing number of genes associated with 46,XY GD, we believe that single gene sequencing is no longer efficient or cost effective (discussed in the Section “High Throughput Sequencing in the Diagnosis of Disorders of Sex Determination”). In addition, whole-exome sequencing can also detect deletions and duplications at the level of exons, although the gold standard for larger deletions/duplications remains comparative genomic hybridization (CGH). CGH can alternatively be used to detect relatively small deletions or duplications in or around any of the known sex-determining genes. As such, noncoding duplications and deletions have also been identified surrounding SOX9 , GATA-4 , and SOX3. Therefore, in patients with 46,XY GD we recommend whole-exome sequencing to identify the causative genetic diagnosis for patients with DSD.
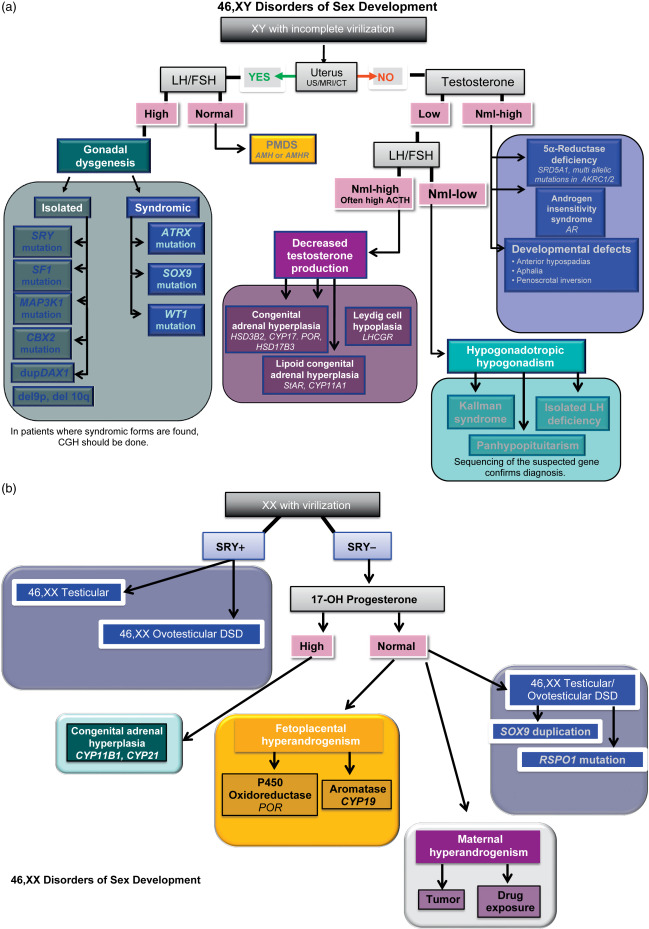
Ultimately, the definitive diagnosis to determine whether one has complete, partial, or mixed GD is through biopsy of the gonads. While this is a useful endeavor, it may not provide significant insight into the biological or genetic basis of disease or any predictive value outside of fertility and gonadoblastoma risk. Bilateral streak gonads are seen in pure GD, bilateral dysgenic testes in partial GD, and a unilateral streak gonad with contralateral normal to dysgenic testis in mixed 46,XY GD. The risk of otherwise undetected gonadoblastoma in 46,XY GD patients is high, and prophylactic or therapeutic gonadectomy is therefore often indicated when 46,XY GD is diagnosed.
46,XX Disorders
Testicular and Ovotesticular DSD
There is a range of 46,XX individuals in which the gonads are testicular or ovotesticular. Patients with 46,XX testicular or ovotesticular DSD are likely to be part of the same phenotypic spectrum. Ultimately, differentiating the two can only be done with gonadal biopsy, which is not always performed; therefore, an exact diagnosis cannot always be made.
In 46,XX testicular DSD, with an approximate incidence of 1:20,000, varying amounts of testicular (but no ovarian) tissue are present. About 85% of 46,XX testicular DSD patients are phenotypic males with unambiguous male genitalia at birth, and are not diagnosed until puberty fails to proceed normally. There can be similarities with Klinefelter (46,XXY), including diminished secondary sex development, gynecomastia, small testes, and azoospermia. However, in contrast, cognitive ability is normal, stature is normal to short, and ejaculation and sexual function are normal. Approximately 85% of these patients carry an Xp:Yp translocation that contains SRY . The remaining 15% of 46,XX testicular DSD individuals have ambiguous genitalia, and ectopic presence of SRY accounts for only a minority of these cases. In addition, disruption of R-spondin1 ( RSPO1 ), a female-determining gene, results in a recessive syndrome that includes complete 46,XX sex reversal ( Table 19.1 ), but the etiology of many 46,XX testicular DSD patients remains unclear.
In 46,XX ovotesticular DSD (formerly true hermaphroditism), both ovarian and testicular tissues (defined by the presence of follicles and seminiferous tubules, respectively) are present. Approximately 50% of cases have an ovary and an ovotestis; 30% have two ovotestes; and 20% have an ovary and a testis, resulting in variable development of internal and external genitalia. The ipsilateral predominance of ovarian or testicular tissue typically correlates with the location of each gonad and the degree of Müllerian- and/or Wolffian-derived development on that side. About 7–10% of ovotesticular DSD are actually 46,XY; mutations in SRY have been found in two such patients. An estimated 30–33% of ovotesticular DSD are mosaics – including 46,XX/46,XY – with some Yp involvement. The majority – approximately 60% – of ovotesticular DSD are 46,XX. Of these, a minority are SRY-positive, placing these patients on a phenotypic continuum with 46,XX testicular DSD. As with some 46,XX testicular DSD patients, the etiology can be an Xp:Yp translocation. There is evidence based on genotyping of peripheral lymphocytes that translocation of Yp onto the active X in a majority of cells can result in 46,XX testicular DSD, while translocation onto the inactive X in most cells may cause inactivation to spread into the Yp region and result in the more ambiguous phenotype of 46,XX ovotesticular DSD. Syndromic 46,XX ovotesticular, as well as testicular, DSD has been attributed to a mutation in RSPO1.
A duplication containing the male-determining gene Sox9 has been implicated in 46,XX testicular and ovotesticular DSD in one patient and, more recently, duplications and deletions in the regulatory regions of SOX9 and other SOX genes leading to aberrant upregulation of SOX9 or other SOX genes is sufficient to promote testis formation in an 46,XX individual.
Presentation and Diagnosis
Most patients with 46,XX testicular DSD have “de la Chapelle syndrome” with an unambiguous male phenotype that is unassociated with other disease diagnoses, and present in adolescence with delayed puberty or infertility. Although patients do occasionally present in childhood with undervirilized genitalia, SRY (found in the majority of 46,XX testicular DSD) is often correlated with relatively high virilization of the genitals, and fewer than 20% of 46,XX testicular DSD patients present as preadolescents. In 46,XX testicular DSD, analysis of the semen typically shows a normal volume with azoospermia. Karyotyping of peripheral blood cells shows 46,XX in the majority of cases. FISH for SRY is positive in 90% of patients; if negative, FISH for SOX9 microduplications can be performed, and CGH can be done if molecular etiology is still needed. 46,XX testicular DSD patients may have hypergonadotropic hypogonadism with elevated FSH and LH, decreased T, dihydrotestosterone (DHT), less than twofold increase in response to the human chorionic gonadotropin (hCG) stimulation test, and no uterus, as determined by pelvic ultrasound (refer to Table 19.2 and Fig. 19.2 b).
The majority of patients with ovotesticular DSD present with ambiguous genitalia in infancy or early childhood. Unlike patients with 46,XX testicular DSD, those with ovotesticular DSD often have some ovarian function. Hormonal levels ( Table 19.2 ) correlate with the relative amounts of testicular and ovarian tissue; FSH, LH, E 2 , T, DHT, and Δ4A can be in the normal female range. Hormonal levels in turn correlate with the degree of genital ambiguity. The karyotype for the majority of patients with ovotesticular DSD will be 46,XX, but approximately one-third will be mosaic (including 46,XX/46,XY), or, rarely, 46,XY.
Syndromic effects of SOX9 duplications or RSPO1 mutations ( Table 19.1 ) will suggest appropriate tests for these criteria, but results will not differentiate between 46,XX testicular and ovotesticular DSD. The biochemical marker for Sertoli cells is a serum AMH level greater than 75 nmol/L, which is unequivocal evidence of functional testicular tissue, and suggests either 46,XX testicular or ovotesticular DSD, but again does not distinguish between the two. Ultimately, a thorough biopsy of both gonads remains the basis for a definitive differential diagnosis: the presence of any ovarian tissue distinguishes ovotesticular from 46,XX testicular DSD. In addition, as SRY expression has been detected in gonads of patients who are otherwise SRY negative, tests for karyotype, cryptic mosaicism, and SRY can be performed in gonadal tissue, if indicated.
Sex Chromosome Disorders
Sex chromosome DSD results from having an abnormal number of sex chromosomes. Errors in paternal meiosis are a cause in a majority of these disorders.
Turner Syndrome
Turner syndrome is a disorder affecting females in which all or a part of one X chromosome is missing. The majority of Turner syndrome patients are 45,X; the rest are 46,XX with X chromosome deletions or mosaics with various combinations of sex chromosome number or content. 45,X Turner syndrome is relatively common in the population, and while less than 3% of Turner zygotes survive to term, approximately one in 2000 newborn phenotypic females are 45,X.
Most Turner females are short in stature due to lack of one copy of the homeobox gene SHOX , which is located in the pseudoautosomal region. They possess streak gonads, with fewer and poorly developed follicles in utero. Given the reduced number of follicles, Turner patients have less estrogen secretion, resulting in delayed puberty or primary amenorrhea.
Presentation and Diagnosis
Turner syndrome should be considered if prenatal ultrasound reveals short femur, total body lymphangiectasia, large septate cystic hygromas, nuchal thickening, and/or cardiac defects. Turner newborns may present with low birth weight, lymphedema of the upper and lower extremities (in 30% of Turner babies), a webbed neck (pterygium colli), and dysmorphic features: low-set prominent ears, low posterior hairline, micrognatia, high-arched palate, epicanthal folds, hypoplastic nail beds, and/or hypoplastic fourth and fifth metacarpals. Turner adolescents present most frequently with short stature, amenorrhea, and lack of secondary sex characteristics, although (depending on the amount of GD) approximately 30% do have some spontaneous puberty.
Additional characteristics of Turner syndrome may include: renal anomalies (incidence between 30% and 50%), increased frequency of cardiovascular disease such as coarctation of the aorta, hearing loss, shield-like chest, and higher carrying angle of the arms (cubitus valgus).
If a patient’s presentation suggests Turner, karyotyping should be done to detect 45,X Turner. If this assay is normal, FISH or CGH should be done to detect possible cryptic deletions in the SHOX -containing pseudoautosomal region of an X chromosome in a 46,XX patient.
Klinefelter Syndrome
Klinefelter syndrome, or 47,XXY, males have a normal number of primordial germ cells in utero , which degenerate through childhood probably due to a fault of communication between Sertoli and germ cells. Roughly 50% of Klinefelter syndrome occurrence is of paternal origin with a possible increase in 46,XY sperm frequency as paternal age increases.
Presentation and Diagnosis
The phenotype of Klinefelter syndrome is often not obvious and as such remains primarily underdiagnosed in the general population. Behavioral disorders, abnormally small testes and legs disproportionately long compared to upper extremities, may be seen in Klinefelter boys. A patient’s IQ may be somewhat lower than siblings, though typically still in the normal range. In adolescence, most Klinefelter patients present with small, firm testes and hypogonadism with some degree of androgen deficiency. Later, males may present at infertility centers with azoospermia.
Diagnosis of Klinefelter syndrome is performed by karyotype of lymphocytes. Some mosaic cases will only be detected by karyotype of skin fibroblasts and occasionally of testicular biopsy.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


