The nomenclature, Disorders of Sex Development (DSDs), replaces the term “intersex,”1,2 to encompass various congenital conditions in which there is inconsistency between chromosomal, gonadal, and/or anatomical sex. Conditions with ambiguous external genitalia are included, encompassing atypical genital development. While this system (Table 6-1) allows the incorporation of new genetic and other information, the primary clinical advantage is a guideline for early diagnostic evaluation, approaching by phenotype and karyotype, to guide further etiological evaluation and management. This classification of diagnostic categories (Table 6-2) is limited because some conditions fail to fit into a single specific diagnostic category or fit into more than one category; for example, forms of gonadal dysgenesis may have a 46,XY, 46,XX, or 45,X/46,XY karyotype. This schema includes the genetic cause when known and allows for phenotypic variation within etiological categories. DSDs primarily include those conditions impacting reproductive or sexual function, while including variations of normal development such as mild hypospadias and idiopathic clitoromegaly. Even though a karyotype is used for initial categorization, the ideal current approach involves specific molecular etiologies, as genes are more specific, and would avoid the perception that sex is determined by chromosomes. For example, the presence of a Y chromosome may not be associated with consistent findings regarding factors important in determining sex assignment or assessing outcome, including fetal androgen exposure, genital masculinization, or germ cell development.
| Sex Chromosome DSD | 46,XY DSD | 46,XX DSD |
|---|---|---|
| 45,X Turner syndrome | Aberrant testicular development | Aberrant ovarian development |
| 47,XXY Klinefelter syndrome | Disorders of androgen synthesis or action | Disorders of androgen excess |
| 45,X/46,XY Mixed gonadal dysgenesis | Disorders of MIH synthesis or action | Vaginal or uterine atresia |
| 46,XX/46,XY Chimeric, ovotesticular | Other | Other |
| A. Sex chromosome DSD |
| 1. 45,X—Turner syndrome and variants |
| 2. 47,XXY—Klinefelter syndrome and variants |
| 3. 45,X/46,XY—Mixed gonadal dysgenesis, ovotesticular DSD |
| 4. 46,XX/46,XY (Chimeric, ovotesticular DSD) |
| B. XY DSD |
| 1. Disorders of testicular development |
| a. Complete gonadal dysgenesis (Swyer syndrome) |
| b. Partial gonadal dysgenesis |
| c. Gonadal regression syndrome |
| d. Ovotesticular DSD |
| 2. Disorders in androgen synthesis or action |
| a. Androgen biosynthesis defects |
| i. 5α-Reductase deficiency |
| ii. 17-Hydroxysteroid dehydrogenase deficiency |
| iii. StAR mutations |
| b. Defects in androgen action |
| i. Partial androgen insensitivity |
| ii. Complete androgen insensitivity |
| c. LH receptor defects (Leydig cell aplasia/hypoplasia) |
| d. Disorders of AMH (MIH) and AMH receptors (persistent müllerian duct syndrome) |
| 3. General categories |
| a. Hypospadias not associated with hormone defect |
| b. Cloacal exstrophy |
| C. 46,XX DSD |
| 1. Disorders of gonadal (ovarian) development |
| a. Ovotesticular DSD |
| b. SRY+, dup SOX9 testicular DSD |
| c. Gonadal dysgenesis |
| 2. Androgen excess |
| a. Fetal adrenal |
| i. 21-Hydroxylase deficiency |
| ii. 11-Hydroxylase deficiency |
| b. Fetoplacental |
| i. Aromatase deficiency |
| ii. Cytochrome P450 oxidoreductase deficiency |
| 3. General categories |
| a. Cloacal exstrophy |
| b. Vaginal atresia (Mayer-Rokitansky-Küster-Hauser syndrome) |
| c. MURCS association (müllerian hypoplasia/aplasia, renal agenesis, and cervicothoracic somite abnormalities) |
Accurate estimates regarding the incidence of DSDs are not available because of the multiple and rare etiologies. The most common cause of a DSD, classicadrenal hyperplasia (CAH) secondary to 21-hydroxylasedeficiency, occurs in 1:10,000-1:18,000 births as determined by newborn screening data, with considerable variation among ethnic/racial backgrounds related to mutation frequency. Deficiency of 11-hydroxylase is generally rare, although the incidence is high among Moroccan Jews (∼1 in 6000). The estimated incidence of 17-hydroxysteroid dehydrogenase deficiency is 1 in 147,000. The Smith-Lemli-Opitz syndrome, which is associated with multiple congenital abnormalities, including urogenital anomalies, occurs with an estimated incidence of 1 in 20,000 to 60,000 live births. There are no estimated incidences of most rare DSDs.
Differentiation of the reproductive/sexual system can be divided into the components outlined in Table 6-3.
| Precursor | Male | Female |
|---|---|---|
| 1. Internal duct systems | ||
| a. Mesonephric (wolffian) | Epididymis, vas deferens, and seminal vesicles | Regression |
| b. Paramesonephric (müllerian) | Regression Prostatic utricle | Fallopian tubes, uterus, and upper part of vagina |
| 2. Urogenital sinus | (Urethra) | Lower portion of vagina (and urethra) |
| 3. External genitalia | ||
| a. Genital tubercle | Corpora cavernosa of penis | Clitoris |
| b. Labioscrotal folds | Scrotum | Labia majora |
| c. Labiourethral folds | Penile urethra, glans, and corpus spongiosum | Labia minora |
| 4. Undifferentiated gonad | Testis | Ovary |
The paramesonephric/müllerian and mesonephric/wolffian ducts, primordial reproductive structures, develop in both sexes. In fetal testes, Sertoli cells begin to secrete anti-müllerian hormone (AMH) (also known as müllerian-inhibiting hormone [MIH] or müllerian-inhibiting substance [MIS]) by the seventh week of gestation. This hormone triggers side-specific regression of müllerian duct development. Subsequently, testosterone secreted by testicular Leydig cells, under the drive of placental human chorionic gonadotropin (hCG) before the fetal pituitary begins to secrete luteinizing hormone (LH) around the 12th week of gestation, promotes side-specific wolffian duct development into the epididymis, vas deferens, and seminal vesicle. The lack of both of these testicular hormones in the normal female fetus allows müllerian duct maturation into fallopian tubes, uterus, cervix, and upper vagina, without wolffian duct development.
Primordial bipotential genital structures, the genital tubercle, urethral folds, and labioscrotal swellings, differentiate into male or female external genitalia. The enzyme, 5α-reductase type 2 (hereafter referred to as 5α-reductase), expressed in these structures in the male fetus, converts testosterone, secreted by the fetal testes, into dihydrotestosterone (DHT). DHT, produced in sufficient amounts from testosterone even from a single functioning testis, induces male external genital differentiation. In the normal human male fetus, a cylindrical phallus approximating 2 mm in length with genital swellings develops by 9 weeks of gestation. The developing penis is not larger than the clitoris at 12 weeks although sonography can differentiate the male from the female phallus based on its angle. By 14 weeks of gestation, the urethral folds fuse to form the cavernous urethra and corpus spongiosum. The genital tubercle develops into the corpora cavernosa of the penis, and the labioscrotal folds fuse from posterior to anterior to form the scrotum. By 14 weeks, the external genitalia are clearly masculine except for testicular descent. Fetal phallic growth after genital differentiation is a result of fetal pituitary LH stimulation of testosterone production by testicular Leydig cells. Anogenital distance (AGD), measured from the anus to the most posterior edge of the labia or scrotum, is an index of extent of androgen exposure occurring during a masculinization programming window, likely at the onset of fetal genital differentiation, based on data from rodents.3 While such a masculinization programming window suggests a regulated limit for genital growth, adequate androgen exposure subsequently is crucial for maximal growth. Hence, for the clinician, it is unclear to what extent treatment with androgen will increase penile length. Though the AGD may be useful when assessing male physical and physiologic development at any age because reduced AGD distance suggests diminished androgen exposure during the aforementioned masculinization programming window among males, the clinical application is limited. Initial assessment may still involve androgen stimulation to assess responsiveness. A diminished AGD for age has been associated with hypospadias or poorer sperm quality, or exposure to endocrine disruptor chemicals.
Deficient testosterone production, inadequate testosterone action, or other factors result in incomplete fusion of the genital components as illustrated in Figure 6-1. This figure depicts genitalia along the spectrum from female to male as may occur in a 46,XX fetus with excessive androgen stimulation or in a 46,XY fetus with deficient androgen exposure and/or response. However, the size and extent of fusion of phallic development do not necessarily correlate with the extent of labioscrotal fusion, so each should be described separately. The high prevalence of hypospadias in boys suggests that urethral fusion is a delicate and finely regulated process that may be impacted by numerous other factors in addition to androgen. The degree of fusion of the urethra may extend further toward the tip than fusion of the underlying spongiosum, creating a condition that makes surgical repair difficult.
Figure 6-1
Shows lateral and en face views of external genitalia of the fully differentiated female on the left, increasing amounts of virilization (excessive for a female and inadequate for a male) going from left to right, with complete male differentiation on the right. (Reproduced with permission from Ogilvy-Stuart AL, Brain CE. Early assessment of ambiguous genitalia. Arch Dis Child. 2004;May;89(5):401-407.)
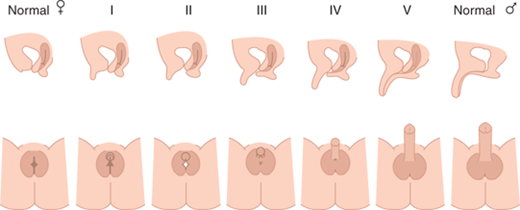
The critical role of DHT to promote full differentiation of male external genitalia is illustrated by the incomplete differentiation of male external genitalia among boys with 5α-reductase deficiency. Affected boys have normally functioning testes and normal testosterone production, but they are unable to convert testosterone to DHT within external genital structures and the prostate during fetal life. However, increased androgen exposure at puberty typically results in further virilization. Such undervirilization at birth has led to a female sex of rearing, particularly if the diagnosis is not recognized, whereas virilization at puberty has led to gender reassignment. In the modern era, hopefully, the diagnosis is made in a timely fashion leading to a male sex assignment.
In the absence of androgens or androgen action, the urethral folds and labioscrotal swellings in the 46,XX (or 46,XY) fetus remain unfused and develop into the labia minora and labia majora, respectively. The genital tubercle forms the clitoris, and canalization of the vaginal plate creates the lower portion of the vagina. By 11 weeks of gestation, the clitoris is prominent and the lateral boundaries of the urogenital sulcus have separated. The separation of vagina and urethra, which is not hormone dependent, is completed by 12 weeks. However, high levels of androgen prevent this separation as evidenced by high insertion of the urethra into the vagina that may occur with CAH. At 20 weeks of gestation, normal female differentiation results in minimal clitoral growth and well-defined labia majora, but as yet relatively hypoplastic labia minora with separate vaginal and urethral perineal openings. Excessive androgen stimulation from any source in the female fetus during the period of genital differentiation prior to 12 weeks of gestation results in labial fusion and phallic urethral development, apparently related to extent of androgen exposure (see Figure 6-1). Later during gestation androgen excess cannot change differentiation, but it results in excessive clitoral growth and scrotalization of the unfused labial folds. The gestational timing of androgen excess can thus be estimated by the extent of masculinization (differentiation) and virilization (growth excess) of the genitalia.
The urogenital ridges, the primordial structures that form the gonads, reproductive tract, kidneys, and adrenal cortices, are present by 6 weeks of gestation, are outgrowths of coelomic epithelium (mesothelium). Proximity to the mesonephros, the source of endothelial cells, is necessary for gonadal differentiation. The primitive gonad in the urogenital ridge has a cortex and a medulla. The medullary portion begins to differentiate into testicular tissue around the seventh week of gestation, earlier than ovarian development from the primitive gonadal cortex.
SRY (sex-determining region on the Y chromosome) is a nuclear high-mobility group (HMG) domain protein expressed in pre-Sertoli cells where it triggers a molecular switch to initiate the process of male sexual differentiation. In fact, the first evidence of testicular differentiation, induced by the SRY protein, is the appearance of primitive Sertoli cells at 6 to 7 weeks of gestation. This is followed by migration of endothelial cells and the appearance of peritubular myoid cells, Leydig cells, and germ cells. Interaction of primitive Sertoli cells and endothelial cells results in testicular cord development, which become seminiferous tubules containing mature Sertoli and germ cells. Cell proliferation rates increase, and germ cells enter a state of mitotic arrest.
Germ cell meiosis in the ovary begins at 10 to 11 weeks of gestation. Over the next 10 weeks, after the appearance of oögonia, structured follicles are present. Primary follicles, derived from granulosa cell precursors, develop adjacent to the primary oöcyte, becoming evident at 20 weeks of gestation. As many as 7 million oöcytes present at mid-gestation begin to degenerate, so approximately 2 million are present at birth. Such follicular atresia appears to be accelerated in the streak gonads of fetuses with X monosomy.
The sequence of the development of the components of the reproductive system development (Figure 6-2) involves differentiation of the bipotential primordial structures discussed earlier.
Figure 6-2
Shows the age (in weeks of gestation) of the onset of anatomical changes and differentiation allowing for comparison of relative age of onset. Male differentiation is shown above the line for each primordial structure with female below.53 (Reproduced with permission White and Speiser.)
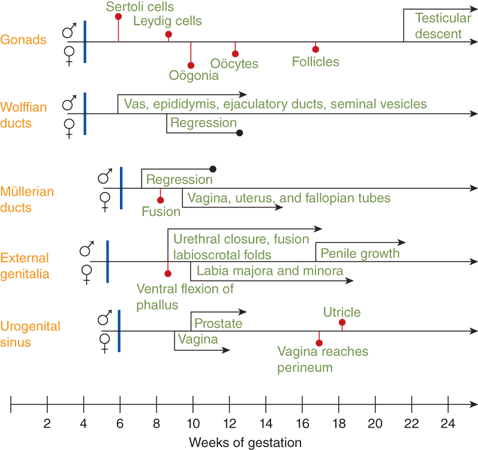
Differentiation of the testis with discernible Sertoli and Leydig cell lines beginning around the seventh to eighth week of gestation precedes ovarian differentiation in which oögonia do not appear until the 10th week of gestation. Secretion of testosterone by fetal Leydig cells begins concomitantly with differentiation of the wolffian ducts; müllerian duct regression in the male is evident slightly later. Male external genital differentiation begins around the eighth week of gestation and is completed by the 12th to 14th week. Virilization continues with penile growth and, in the presence of excessive androgen in the female fetus, clitoral enlargement. Vaginal development occurs during this period with the lower two-thirds arising from an invagination of the epidermal layers and merging with the upper third that develops from a downward extension of the müllerian ducts.
Genes known to be involved in the differentiation of the gonads, external genitalia, and internal genitalia are depicted in Figure 6-3.
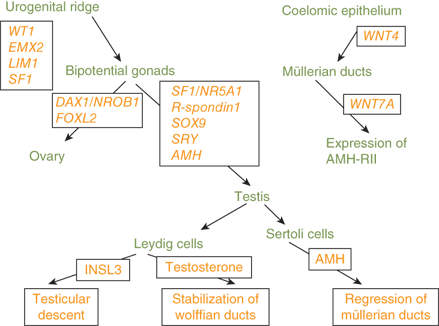
The SRY gene encodes a transcription factor that initiates the formation of the testes. Subsequently, the SOX9 gene, which is necessary for Sertoli-cell differentiation and type 2 collagen production, is expressed in the testes.4 Mutations of SOX9 are associated with sex reversal. Haploinsufficiency of SOX9 results in campomelic dysplasia, a disorder showing skeletal dysplasia that is associated with sex reversal in the majority of affected XY individuals (XY sex reversal). Familial campomelic dysplasia associated with a deletion upstream of SOX9 has been described in a mother and 46,XY child with female external genitalia, normal uterus, and streak gonads.5 Duplication of SOX9 is a cause of a male phenotype and an XX karyotype (XX sex reversal).
Other molecular factors involved in gonadal differentiation include steroidogenic factor-1 (SF1), a transcription factor necessary for steroidogenesis, male sexual differentiation, and fertility. Mutations of the SF1/NR5A1 gene have been identified in patients with agonadism, adrenal hypoplasia, hypogonadotropic hypogonadism, cryptorchidism, micropenis, and XY sex reversal.6
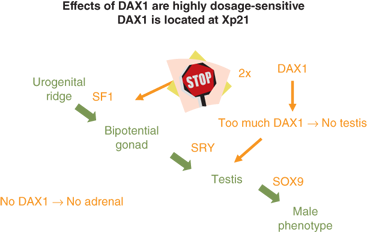
A gonad-specific transcription factor upregulated in the ovary called DAX1 (Dosage-sensitive Adrenal hypoplasia congenita region on the X chromosome) that is encoded by the DAX1/NROB1 gene is not required for normal testicular development, but it appears to function as an antitestis factor in the XX ovary (Figure 6-4). Duplication of DAX1 in the 46,XY fetus is thought to repress SRY resulting in XY sex reversal, and DAX1/NROB1 mutations are associated with a syndrome of adrenal hypoplasia and hypogonadotropic hypogonadism (adrenal hypoplasia congenita [AHC]).
Figure 6-4
Depicts the normal developmental sequence along with the impact of duplication or deletion of DAX1/NROB1 on testicular and adrenal development.54 (Adapted, with permission, from Meeks et al.)
Thus, a threshold level of SRY activity is necessary for male sexual differentiation. Sequential expression of several other genes, including SRY-related HMG box-containing-9 (SOX9), MIH, DAX1/NROB1, steroidogenic factor-1 (SF1/NR5A1), Wilms tumor 1 (WT1), GATA-binding-4 (GATA4), desert hedgehog (DHH), patched (PTC), wingless-related mouse mammary tumor virus (MMTV) integration site 4 (WNT4), and WNT7a, are required for normal male sexual differentiation. Cells, mostly endothelial in origin, migrate from the mesonephros and interact with the pre-Sertoli cells to promote development of the testicular cords.
Although it has generally been considered that ovarian differentiation is constitutive, evidence suggests that specific genes are required for normal ovarian development. Primordial germ cells migrate from their origin in the hindgut and colonize in the genital ridges under the influence of fragilis (interferon-induced transmembrane) proteins 1 and 3 (IFITM1 and IFITM3), stromal cell-derived factor 1 (SDF1 or CXCL12), and its receptor CXCR4.
Steroidogenic enzyme gene mutations are a frequent cause of genital ambiguity resulting from alterations in sex steroid biosynthesis. These enzymes are expressed in the placenta, gonads, and adrenal cortex. In the 46,XY fetus, specific proteins necessary for testosterone and DHT biosynthesis include SF1, steroidogenic acute regulatory peptide (StAR), 17α-hydroxylase/17,20-lyase (CYP17), 3β-hydroxysteroid dehydrogenase type 2 (HSD3B2), 17α-hydroxysteroid dehydrogenase type 3 (HSD17B3), P450-oxidoreductase (hereafter referred to as oxidoreductase), and 5α-reductase type 2 (SRD5A2) (Figure 6-5). Defective testosterone biosynthesis can lead to ambiguous genitalia in the 46,XY fetus.
Figure 6-5
Depicts the steroidogenic pathways of the adrenal cortex, ovary, and testis. Common pathways of both the adrenals and gonads are shown across the top of the figure. The steroids and enzymes present in the adrenal cortex are within the solid line, whereas enzymes and steroids not present in the adrenal cortex, but unique to the ovary and testis, are shown within the broken lines. CYP11B1, 11β-hydroxylase type 1; CYP11B2, 11β-hydroxylase type 2; CYP17, 17α-hydroxylase/17,20-lyase; CYP19, aromatase; CYP21, 21α-hydroxylase; HSD3B2, 3β-hydroxysteroid dehydrogenase type 2; HSD17B1, 17α-hydroxysteroid dehydrogenase type 1; HSD17B3, 17α-hydroxysteroid dehydrogenase type 3; SRD5A2, 5α-reductase type 2.
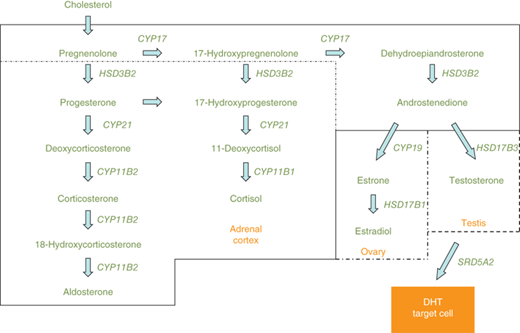
Steroidogenic pathways of the adrenal cortex, ovary, and testis are depicted in Figure 6-5. Differences in enzyme expression are indicated by the specific boxes showing androstenedione as the endpoint of sex steroid synthesis in the adrenal (zona reticularis), testosterone in the testis, and estradiol in the ovary. Other steroid endpoints in the adrenal cortex include aldosterone (zona glomerulosa) and cortisol (zona fasciculata).
21-Hydroxylase deficiency caused by mutations in the 21-hydroxylase (CYP21A2) gene is the most common cause of CAH. This autosomal recessive disorder is responsible for more than 95% of the cases of 46,XX genital ambiguity. Deficient 21-hydroxylase activity results in decreased cortisol biosynthesis, secondary to insufficient conversion of 17-hydroxyprogesterone (17-OHP) to 11-deoxycortisolin the zona fasciculata, and more variable degrees of decreased aldosterone biosynthesis from impaired conversion of progesterone to deoxycorticosterone in the zona glomerulosa. The loss of negative feedback inhibition from cortisol leads to increased adrenocorticotropic hormone (ACTH) secretion with subsequent increased adrenal androgen secretion (Figure 6-6).

46,XX patients with severe 21-hydroxylase deficiency manifest genital ambiguity at birth. Virilization of the external genitalia exists along a spectrum ranging from mild clitoromegaly and mild posterior labial fusion to, although rare, a genital appearance consistent with a normal male with bilateral cryptorchidism. Clitoromegaly results from any source of excessive fetal androgen, and hence the various causes are listed in Box 6-1. Despite the excessive prenatal androgen exposure, affected females have normal internal reproductive anatomy with a uterus, normally positioned ovaries, fallopian tubes, and upper vagina (müllerian derivatives). Because of glucocorticoid and mineralocorticoid deficiencies, severely affected untreated infants develop hyponatremia, hyperkalemia, and dehydration around 7 to 10 days of life. The serum 17-OHP concentrations are usually more than 10,000 ng/dL and are often considerably higher. Serum androstenedione and progesterone concentrations are also elevated, but to a lesser degree than are those of 17-OHP. Measurement of plasma renin activity (PRA) is necessary to assess mineralocorticoid status, although discernment in the neonatal period may be difficult because of normally high levels. Newborn screening programs in all 50 states measure whole-blood 17-OHP concentrations obtained on filter paper with the primary goal of permitting early detection in affected male infants. Newborn screening programs generally do not detect children with nonclassic adrenal hyperplasia (NCAH), which is not a form of DSD.
Box 6-1.
Clitoromegaly in a 46,XX infant usually results from excess androgen exposure as occurs in patients with:
21-Hydroxylase deficiency
11-Hydroxylase deficiency
3β-Hydroxysteroid dehydrogenase deficiency
Placental aromatase deficiency
Oxidoreductase deficiency
Ovotesticular DSD
Testicular DSD
Maternal androgen exposure
Also, rarely results from inclusion of other tissues, such as neurofibromatosis
The degree of impairment of 21-hydroxylase activity varies depending on the compromise of enzyme function associated with specific mutations. In general, complete loss-of-function mutations are associated with more severe virilization of the external genitalia in affected females and clinically relevant salt wasting. The simple-virilizing form of CAH is associated with a 1% to 2% level of residual enzyme activity whereas that of NCAH generally has a 20% to 60% level of enzyme activity. Over 100 mutations of the 21-hydroxylase gene, CYP21A2, have been reported. A limited number of mutations account for the majority of the affected alleles, and there appears to be reasonable genotype-phenotype correlation.
CAH caused by 11β-hydroxylase deficiency is associated with glucocorticoid deficiency, hypertension (excessive concentrations of salt-retaining hormones), and excessive androgen secretion. Curiously, some infants with 11β-hydroxylase deficiency may manifest salt loss and hypotension in the neonatal period. Affected female infants may present with ambiguous genitalia. In contrast to patients with 21-hydroxylase deficiency, those with 11β-hydroxylase deficiency have a hormonal profile with elevated 11-deoxycortisol and relatively less severe elevations in 17-OHP, androstenedione, and testosterone. PRA concentrations are typically, but not invariably, suppressed. Mutations in the 11β-hydroxylase (CYP11B1) gene, located on chromosome 8q22, are causative.
CAH caused by deficiency of 3β-hydroxysteroid dehydrogenase, type 2, may result in genital ambiguity in both 46,XX and 46,XY fetuses. In this autosomal recessive disorder, increased dehydroepiandrosterone (DHEA) synthesis leads to virilization in the 46,XX fetus, whereas deficient testosterone production results in undervirilization in the 46,XY fetus. The condition is characterized by elevated serum concentrations of the Δ5 steroids (pregnenolone, 17-hydroxypregnenolone, and DHEA) because of a reduced ability to convert them to their corresponding Δ4 steroids (progesterone, 17-OHP, and androstenedione) (see Figure 6-5). Impaired biosynthesis of mineralocorticoids, glucocorticoids, and sex steroids is associated with acute adrenal insufficiency that may occur in the newborn period. Mutations in the HSD3B2 gene, located on chromosome 1p13.1 and expressed in the adrenal cortex and gonads, have been documented in affected individuals.
Swiss families previously reported to have isolated 17,20-lyase deficiency have been studied7 and found to have a normal CYP17A1 gene, but instead to harbor mutations in the 3α-hydroxysteroid dehydrogenase type III (AKR1C2) and another in the 3α-HSD gene (AKR1C4). This identifies alternative pathways for DHT synthesis (bypassing testosterone), in addition to the classically described pathways that work through testosterone, leading to the formation of the same end products. This finding suggests that both the classic and alternative “backdoor” pathways are necessary for normal male external genital development.
The enzyme, aromatase, catalyzes the biosynthesis of estrogens from androgens. Aromatase deficiency is a rare autosomal recessive disorder in which inactivating mutations of the aromatase (CYP19A1) gene cause increased serum androgen levels. Because this enzyme promotes estrogen biosynthesis, there are distinct phenotypes in males and females.8 Aromatase is active in the placenta, where it normally protects the mother from the potential virilizing effects of the fetal androgens (predominantly DHEA and DHEAS). In placental aromatase deficiency, 46,XX fetuses are virilized and manifest variable degrees of labioscrotal fusion, clitoromegaly, and perineoscrotal hypospadias. Often, there is a history of progressive maternal virilization during pregnancycharacterized by acne, hirsutism, clitoral hypertrophy, and frontal balding. During the pregnancy, maternal concentrations of testosterone, DHT, and androstenedione are elevated and decline rapidly after delivery. Affected 46,XX children have delayed pubertal development secondary to hypergonadotropic hypogonadism with poor breast development, primary amenorrhea, and cystic ovaries. Affected 46,XY infants have normal internal and external genital development but, at the age of puberty, may have tall stature and continued growth (asymmetrical with relatively long legs) because estrogen is required to fuse the growth plates of the long bones. The estrogen deficiency found in men with aromatase deficiency is associated with abdominal obesity, insulin resistance, dyslipidemia, and relativeinfertility.
Another disorder affecting steroidogenesis is characterized by decreased 17α-hydroxylase and 21-hydroxylase activities. The phenotype includes genital ambiguity in males and females, craniosynostosis, mid-face hypoplasia, radiohumeral synostosis, and glucocorticoid deficiency. The skeletal manifestations are reminiscent of those seen in the Antley-Bixler syndrome, an autosomal dominant disorder caused by mutations in the fibroblast growth factor receptor-2 (FGFR2) gene. Affected males may be undervirilized whereas females may be virilized. Signs of androgen excess, including acne, hirsutism, and clitoromegaly, may develop during pregnancy in mothers carrying affected fetuses. Typically, postnatal virilization does not occur. The 17-OHP levels are elevated, sex steroid levels are low, and mineralocorticoid concentrations are normal.
This disorder is caused by loss-of-function mutations in the cytochrome P450 oxidoreductase (POR) gene that is located on chromosome 7q11-12. This gene encodes a protein that functions as a cofactor for several steroidogenic enzymes.9 It has been suggested that an alternative pathway of androgen biosynthesis, with conversion of 17-OHP to 5α-pregnane-3α,17α-dio-20-one to androstenedione, may be seen in this disorder.10
The 17β-hydroxysteroid dehydrogenase type 3 (HSD17B3) gene is expressed in the Leydig cells of the testes where it catalyzes conversion of androstenedione to testosterone (Figure 6-5). Loss-of-function mutations in this gene are inherited in an autosomal recessive manner and result in testosterone deficiency and undervirilization of 46,XY fetuses. In some instances, affected males are thought to be females at birth. The external genital phenotype in the 46,XY infant with 17β-hydroxysteroid dehydrogenase deficiency may range from a completely female (see Figure 6-1) appearance with a blind-ending vaginal pouch to variable degrees of genital ambiguity with gonads palpable in the labioscrotal folds, labioscrotal fusion, and hypospadias.11 Internal male reproductive structures derived from the wolffian ducts are usually present. Underproduction of androgen in females with this mutation appears to confer no obvious phenotype.
Elevated basal or hCG-stimulated androstenedione-to-testosterone ratios are consistent with this enzyme defect. When LH and FSH secretion increases during puberty, virilization may occur because of conversion of androstenedione to testosterone by the other unaffected isoforms of 17β-hydroxysteroid dehydrogenase present in extratesticular tissue. Affected children raised as females have been reported to change gender assignment at puberty. Thus, male gender assignment should be made in infancy for affected children. Breast development may result from increased conversion of androstenedione to estrone.
The 5α-reductase type 2 (SRD5A2) gene is expressed in androgen target tissues such as the prostate and the primordial external genital tissue. This enzyme converts testosterone to DHT (see Figure 6-7), the hormone responsible for virilization of the external genitalia. The external genitalia of affected males are characterized by a small phallus, a urogenital sinus with perineal hypospadias, and a blind vaginal pouch. Internally, male (wolffian) structures are partially differentiated and female (müllerian) ducts are absent. Serum testosterone levels may be elevated, whereas DHT levels are typically low. Elevated basal or hCG-stimulated testosterone-to-DHT ratios (< 10 in normal and > 20 in affected) or diagnostic urinary Δ4/Δ5 steroid profiles are consistent with the diagnosis of 5α-reductase deficiency. In patients whose testicles are left intact, significant virilization often begins during puberty with development of penile growth, male physique, and voice change. In general, male ducts are inadequately developed to deliver mature sperm. Although this autosomal recessive condition is rare, there are clusters of affected individuals in the Dominican Republic, Papua New Guinea, Turkey, and the Middle East.12
Congenital lipoid adrenal hyperplasia (Box 6-2) is an autosomal recessive disorder caused by loss-of-function mutations in the steroidogenic acute regulatory protein (StAR) gene. The StAR protein functions to move cholesterol from the outer to inner mitochondrial membrane,13 which, if absent or deficient, results in impaired conversion of cholesterol to pregnenolone, the first step in steroid synthesis.
Box 6-2. Congenital Lipoid Adrenal Hyperplasia
Consequence of a “two-hit mechanism”
Autosomal recessive disorder due to inactivating mutations in the steroidogenic acute regulatory protein (StAR) gene. The consequence is impaired transport of cholesterol into mitochondria leading to loss of ability to synthesize glucocorticoids, mineralocorticoids, and sex steroids.
Associated with accumulation of cholesterol esters and other cholesterol metabolites that alter cell cytostructure, leading to cell destruction.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


