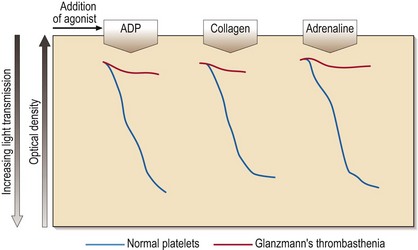
35 Platelet aggregation studies assess the ability of platelets to aggregate in response to the addition of a variety of agonists (e.g. ADP, adrenaline (epinephrine), collagen, arachidonic acid, ristocetin). The tracings produced (Fig 35.1) require expert interpretation. The methodology remains the gold standard with the response to agonists giving characteristic patterns in inherited disorders. Other tests of platelet function include flow cytometry for the quantitation of glycoprotein receptor density, and the measurement of total and/or released adenine nucleotides. The latter tests may confirm the findings from platelet aggregation studies (e.g. in Bernard–Soulier syndrome) or reveal abnormalities where aggregation studies are normal or equivocal (e.g. in a storage pool disease or release defect). This rare autosomal recessive disease is also caused by loss or dysfunction of a platelet glycoprotein complex – GP IIb/IIIa. This normally acts as a receptor for adhesive proteins such as fibrinogen and von Willebrand factor. Platelet numbers and morphology are normal but the platelets fail to aggregate with all agonists except ristocetin (see Fig 35.1). Clinical manifestations are variable but there is typically onset in the neonatal period and subsequent cutaneous and gastrointestinal bleeding, and menorrhagia. Platelet transfusions are indicated where local haemostatic measures fail. If there is platelet refractoriness, recombinant factor VIIa can be used.
Disorders of platelet function and vascular purpuras
Laboratory testing of platelet function
Inherited disorders of platelet function
Glanzmann’s thrombasthenia
Disorders of platelet function and vascular purpuras