Acknowledgment
In memory of Frank B. Diamond, Jr, MD.
Disorders of calcium, magnesium, and phosphate metabolism and of bone formation, accrual, and maintenance during the first 2 decades of life result from suboptimal ingestion, absorption, or retention of constituent nutrients, abnormal vitamin D metabolism or bioactivity, disorders of parathyroid hormone (PTH) synthesis, secretion, or action, and intrinsic aberrations in cartilage and bone cells. The origins of these illnesses may be intrinsic because of pathological variations in the genes controlling these processes or to acquired insults ( Table 20.1 ). Serum concentrations of calcium vary by age. Although serum total and ionized calcium (Ca 2 + ) concentrations generally are intrinsically related, dissociation between these analytes may be observed in patients with hyperproteinemia or hypoproteinemia and at extremes of plasma pH—either alkalosis or acidosis. Hypocalcemia is present when the serum concentrations of Ca 2 + are below, and hypercalcemia is identified by serum values of Ca 2 + above the normal range for age, respectively. For an integrated overview of calcium, mineral, and skeletal homeostasis, the reader is referred to Chapter 9 .
| Gene | Chromosome | OMIM | Disease | OMIM |
|---|---|---|---|---|
| ACVR1 | 2q24.1 | 102576 | Fibrodysplasia ossificans progressiva | 135100 |
| AIRE | 21q22.3 | 607358 | Autoimmune polyendocrine syndrome, type I | 240300 |
| ALPL | 1p36.12 | 171760 | Hypophosphatasia, infantile | 241500 |
| Hypophosphatasia, childhood | 241510 | |||
| Hypophosphatasia, adult | 146300 | |||
| AP2S1 | 19q13.31 | 602242 | Hereditary hypocalciuric hypercalcemia 3 | 600740 |
| BAZ1B | 7q11.23 | 605681 | Williams-Beuren syndrome | 194050 |
| BMP1 | 8p21.3 | 112264 | Osteogenesis imperfecta type XIII | 614856 |
| BSND | 1p32.2 | 606412 | Bartter syndrome type 4a | 602522 |
| CA2 | 8q22 | 611492 | Osteopetrosis – renal tubular acidosis | 259730 |
| CASR | 3q13.3-q21 | 601199 | Hereditary hypocalciuric hypercalcemia 1 | 145980 |
| Neonatal severe hyperparathyroidism | 239200 | |||
| Hypercalcemic hypercalciuria | 601199 | |||
| Hypoparathyroidism, familial isolated | 146200 | |||
| Acquired hypocalciuric hypercalcemia | 145980 | |||
| CDC73 | 1q31.2 | 607393 | Familial isolated hypoparathyroidism type 1 | 145000 |
| Hyperparathyroidism – jaw tumor syndrome | 145001 | |||
| CDKN1B | 12p13.1 | 600778 | Multiple endocrine neoplasia, type 4 | 610755 |
| CHD7 | 8q12.2 | 608092 | CHARGE syndrome | 214800 |
| (Coloboma, Heart defect, Choanal atresia Retardation, Genital and Ear anomalies) | ||||
| CDKN1B | 12P13.1 | 600778 | Multiple endocrine neoplasia type 4 | 610755 |
| CLCN5 | Xp11.23 | 300008 | X-linked recessive hypophosphatemic rickets | 300554 |
| Dent disease 1 | 300009 | |||
| Nephrolithiasis, X-linked recessive | 310468 | |||
| CLCN7 | 16p13,3 | 602727 | Osteopetrosis, autosomal recessive Type IV | 611490 |
| Osteopetrosis, autosomal dominant type II | 166600 | |||
| CLDN10 | 13q32.1 | 617579 | HELIX = Hypohidrosis, Electrolyte imbalance, Lacrimal Dysfunction, Ichthyosis, Xerostomia | 617671 |
| CLDN16 | 3q27 | 603959 | Type 3 hypomagnesemia | 248250 |
| CLDN19 | 1p34.2 | 610036 | Type 5 hypomagnesemia, hypercalciuria, visual impairment | 248190 |
| CLCNKB | 1p36.13 | 602023 | Bartter syndrome type 3 | 607364 |
| CNNM2 | 10q24.32 | 607803 | Type 6 hypomagnesemia with normomagnesuria | 613882 |
| COL1A1 | 17q21.31-q22 | 120150 | Osteogenesis imperfecta type I | 166200 |
| Osteogenesis imperfecta type IIA | 166210 | |||
| Osteogenesis imperfecta type III | 259420 | |||
| Osteogenesis imperfecta type IV | 166220 | |||
| COL1A2 | 7q22.1 | 120160 | Osteogenesis imperfecta type IIA | 166210 |
| Osteogenesis imperfecta type III | 259420 | |||
| Osteogenesis imperfecta type IV | 166220 | |||
| CREB3L1 | 11p11.2 | 616215 | Osteogenesis imperfecta type XV1 | 616229 |
| CRTAP | 3p22 | 605497 | Osteogenesis imperfecta type IIB | 610854 |
| Osteogenesis imperfecta type VII | 610682 | |||
| CTSK | 1q21.3 | 601105 | Pycnodysostosis | 265800 |
| CYP2R1 | 11p15.2 | 608713 | Vitamin D hydroxylation-deficient rickets, type 1B (25-Hydroxylase deficiency) | 600081 |
| CYP3A4 | 7q22.1 | 124010 | Vitamin D–dependent rickets type 3 | |
| CYP24A1 | 12q13.2 | 126065 | Infantile hypercalcemia type 1 | 143880 |
| CYP27B1 | 12q14.1 | 609506 | Vitamin D hydroxylation–deficient rickets, type IA (25α-Hydroxyvitamin D-1α-hydroxylase deficiency) | 264700 |
| DMP1 | 4q22.1 | 600980 | Hypophosphatemic rickets, autosomal recessive type 1 | 241520 |
| ELN | 7q13.23 | 120160 | Williams-Beuren syndrome | 194050 |
| ENPP1 | 6q23.2 | 173335 | Hypophosphatemic rickets, autosomal recessive type 2 | 613312 |
| FAM111A | 11q12.1 | 615292 | Kenny-Caffey syndrome 2 | 127000 |
| Gracile bone dysplasia | 602361 | |||
| FAM20C | 7p22.3 | 611061 | Raine syndrome | 259775 |
| FERMT3 | 11q13.1 | 607901 | Osteopetrosis, autosomal recessive | |
| FGF23 | 12p13.3 | 605380 | Hypophosphatemic rickets, autosomal dominant | 193100 |
| Familial tumoral calcinosis | 211900 | |||
| Hyperostosis hyperphosphatemia syndrome | 610233 | |||
| FGFR3 | 4p16.3 | 134934 | Achondroplasia | 100800 |
| FKBP10 | 17q21.2 | 607063 | Osteogenesis imperfecta type XI | 610968 |
| Bruck syndrome 1 | 259450 | |||
| FOXP3 | Xp11.23 | 300292 | Immunodysregulation, polyendocrinopathy, enteropathy (IPEX) | 304790 |
| FXYD2 | 11q23.3 | 601814 | Autosomal dominant hypomagnesemia type 2 with hypocalciuria | 154020 |
| GALNT3 | 2q24-q31 | 601756 | Familial tumoral calcinosis | 211900 |
| Hyperostosis hyperphosphatemia syndrome | 610233 | |||
| GATA3 | 10p13-14 | 131320 | Hypoparathyroidism, sensorineural deafness, renal disease (hypoparathyroidism-deafness-renal dysplasia/Barakat syndrome) | 146255 |
| GCM2 | 6p24.2 | 603716 | Hypoparathyroidism, familial isolated | 146200 |
| GNA11 | 19p13.3 | 139313 | Hereditary hypocalciuric hypercalcemia 2 | 145981 |
| GNAS | 20q13.32 | 139320 | Pseudohypoparathyroidism, type 1A | 103580 |
| Pseudohypoparathyroidism, type 1B | 603233 | |||
| Pseudohypoparathyroidism, type 1C | 612462 | |||
| Pseudopseudohypoparathyroidism | 612463 | |||
| Osseous heteroplasia, progressive | 166350 | |||
| Fibrous dysplasia/McCune-Albright | 174800 | |||
| GNPTAB | 12q23.2 | 607840 | Mucolipidosis type II | 252500 |
| GTF21 | 7q11.23 | 601679 | Williams-Beuren syndrome | 194050 |
| HADHB | 2p23.3 | 143450 | MELAS—Mitochondrial encephalomyopathy, lactic acidosis, stroke, hypoparathyroidism | 540000 |
| HNF1B | 17q12 | 189907 | Hypomagnesemia with maturity onset diabetes of youth and renal cysts | 137920 |
| HNRNPC | 14q11.2 | 164020 | Vitamin D–dependent rickets type 2B | 600785 |
| HRPT2 | 1q24-q31 | 607393 | Familial hyperparathyroidism 2 – jaw tumor syndrome | 145001 |
| IKBKG | Xq28 | 300248 | Osteopetrosis, X-linked | 300301 |
| IFITM5 | 11p15.5 | 6147577 | Osteogenesis imperfecta type V | 610967 |
| KCNA1 | 12p13.32 | 176260 | Hypomagnesemia with myokymia | 160120 |
| KCNJ1 | 11q24 | 600359 | Hypomagnesemia/Antenatal Bartter syndrome type 2 | 600839 |
| KCNJ10 | 1q23.2 | 612780 | Hypomagnesemia/sesame syndrome | 612780 |
| KL | 13q13.1 | 604824 | Familial tumoral calcinosis | 211900 |
| Hypophosphatemia & hyperparathyroidism | 612089 | |||
| LEPRE1 | 1p34 | 610339 | Osteogenesis imperfecta type VIII | 610915 |
| LRP4 | 604270 | 11p11.2 | Sclerosteosis 2 | 614305 |
| LRP5 | 11.13.4 | 603506 | Osteoporosis-pseudoglioma syndrome | 259770 |
| Idiopathic juvenile osteoporosis | 259750 | |||
| High bone mass variation | 601884 | |||
| Autosomal dominant osteopetrosis type I | 607634 | |||
| Van Buchem disease, type 2 | 607636 | |||
| MBTPS2 | Xp22.12 | 300294 | Osteogenesis imperfecta type XIX | 301014 |
| MEN1 | 11q13 | 613733 | Multiple endocrine neoplasia type I | 131100 |
| NEBL | 10p12.31 | 605491 | DiGeorge syndrome type 2 | 605491 |
| Velocardiofacial syndrome complex 2 | ||||
| NPR2 | 9p21-p12 | 108961 | Acromesomelic dysplasia (Maroteaux) | 602875 |
| OSTM1 | 6q21 | 607649 | Autosomal recessive osteopetrosis type V | 259700 |
| PCBD1 | 10q22.1 | 126090 | Renal hypomagnesemia, maturity onset diabetes of the young, type 5 | |
| PDE4D | 5q11.2-q12.1 | 600129 | Acrodysostosis type 2 | 614613 |
| PHEX | Xp22.2-p22.1 | 300550 | Hypophosphatemic rickets, X-linked dominant | 307800 |
| PLEKHM1 | 17q21.3 | 611466 | Autosomal recessive osteopetrosis type VI | 611497 |
| PPIB | 15q21-q22 | 123841 | Osteogenesis imperfecta type IX | 259440 |
| PRKAR1A | 17q24.3 | 188830 | Acrodysostosis type 1 | 101800 |
| PTH | 11p15.3 | 168450 | Hypoparathyroidism, familial isolated | 146200 |
| PTH1R | 3p21.31 | 168468 | Blomstrand osteochondrodysplasia | 215045 |
| Murk-Jansen metaphyseal chondrodysplasia | 156400 | |||
| Enchondromatosis (Ollier disease) | 166000 | |||
| RET | 10q11.2 | 164761 | Multiple endocrine neoplasia type IIA | 171400 |
| Multiple endocrine neoplasia type IIB | 162300 | |||
| Familial medullary carcinoma of thyroid | 155240 | |||
| SAMD9 | 7q21.2 | 610456 | Tumoral calcinosis, normophosphatemic | 610455 |
| SERPINF1 | 17p13.2 | 172860 | Osteogenesis imperfecta gene type VI | 613982 |
| SERPINH1 | 11q13.5 | 600943 | Osteogenesis imperfecta gene type X | 613848 |
| SLC2A2 | 3q26.2 | 138160 | Fanconi-Bickel syndrome | 227810 |
| SLC34A1 | 5q35.3 | 182309 | Autosomal dominant hypophosphatemia with urolithiasis 1 | 612286 |
| Autosomal recessive Fanconi syndrome with hypophosphatemic rickets | 613388 | |||
| Infantile hypercalcemia, type 2 | 616963 | |||
| SLC34A3 | 9q34.3 | 609826 | Hypophosphatemic rickets with hypercalciuria | 241530 |
| SLC9A3R1 | 17q25.1 | 604990 | Autosomal dominant hypophosphatemia with urolithiasis/osteoporosis 2 | 612287 |
| SLC12A1 | 15q21.1 | 600839 | Antenatal Bartter syndrome type 1 | 601678 |
| SLC12A3 | 16q13 | 600968 | Hypomagnesemia/Gitelman syndrome | 263800 |
| SLC34A1 | 5q35 | 182309 | Infantile hypercalcemia 2 | 616963 |
| SLC34A3 | 9q34 | 609826 | Hypophosphatemic rickets with hypercalciuria, hereditary | 241530 |
| SLC7A7 | 14q11.2 | 603593 | Lysinuric protein intolerance | 222700 |
| SNX10 | 7p15.2 | 614780 | Autosomal recessive osteoporosis type VIII | 615085 |
| SOST | 17q12-q21 | 605740 | Sclerosteosis | 269500 |
| Hyperostosis corticalis generalisata (Van Buchem disease type 1) | 239100 | |||
| SOX3 | Xq26.3 | 313430 | Hypoparathyroidism, X-linked | 307700 |
| SP7 | 12q13.13 | 606633 | Osteogenesis imperfecta XII | 613849 |
| SPARC | 5q33.1 | 182120 | Osteogenesis imperfecta XVII | 6616597 |
| STK3 | 11q23.3 | 614766 | Spondyloepimetaphyseal dysplasia, Krakow | 618162 |
| STX16 | 20q13.32 | 603666 | Pseudohypoparathyroidism, type 1B | 603233 |
| TBX1 | 22q11.12 | 602054 | DiGeorge syndrome | 188400 |
| TBCE | 1q42.3 | 604934 | Sanjad-Sakati (HRD) syndrome | 241410 |
| Kenney-Caffey syndrome, type 1 | 244460 | |||
| TCIRG1 | 11q13.2 | 604592 | Autosomal recessive osteopetrosis type I | 259700 |
| TENT5A | 6q14.1 | 611357 | Osteogenesis imperfecta XVIII | 617592 |
| TGFB1 | 19q13.1 | 190180 | Progressive diaphyseal dysplasia | 131300 |
| TMEM38B | 611236 | 9q31.1 | Osteogenesis imperfecta gene type XIV | 615066 |
| TNFRSF11A | 18q22.1 | 603499 | Autosomal recessive osteopetrosis type VII | 612301 |
| Hereditary (familial) expansile | ||||
| polyostotic osteolytic dysplasia | 174810 | |||
| TNFRSF11B | 8q24 | 602643 | Paget disease, juvenile | 239000 |
| TNFSF11 | 13q14.11 | 602642 | Autosomal recessive osteopetrosis type III | 259730 |
| TRPM6 | 9q21.13 | 607009 | Type 1 hypomagnesemia with hypocalcemia | 602014 |
| TRPV6 | 7q34 | 606680 | Transient neonatal hyperparathyroidism | 618188 |
| VDR | 12q13.11 | 601769 | Vitamin D–resistant rickets, type IIA | 277440 |
| WNT1 | 12q13.12 | 164820 | Osteogenesis imperfecta type XV | 615220 |
Hypocalcemia
In children older than 1 year of age, depending on the analytic laboratory hypocalcemia is defined by a decrease in Ca 2 + values below the lower limits of normal for age of 4.64 to 4.80 mg/dL = 1.16 to 1.20 mmol/L; the lower normal range for age of the total calcium concentration is 8.5 to 8.9 mg/dL = 2.20 to 2.3 mmol/L. If the serum concentration of albumin declines by 1 g/dL, the total calcium value will fall by 0.8 mg/dL (0.2 mmol/L), whereas the level of Ca 2 + does not vary. Symptoms of hypocalcemia reflect heightened neuromuscular irritability, such as paresthesias, tetany, carpopedal spasm, laryngospasm, muscular cramps and/or myotonic spasms, and focal or generalized convulsions; physical signs of hypocalcemia include presence of a Chvostek sign (tapping on facial nerve elicits twitching of facial muscles ipsilaterally) and/or a Trousseau sign (carpopedal spasm after maintaining an inflated blood pressure cuff slightly above systolic pressure for 3 minutes). During hypocalcemia the electrocardiographic QT interval is prolonged.
Hypocalcemia in the Neonate and Infant
In utero, fetal concentrations of total and ionized calcium are higher than they are postnatally; total calcium levels exceed maternal values by approximately 2.0 mg/dL = 0.5 mmol/L, because placental calcium transport is stimulated by parathyroid hormone–related protein (PTHrP); in term umbilical cord blood, mean total calcium values are 10 to 11 mg/dL and Ca 2 + levels approximate 6.4 mg/dL = 1.6 mmol/L, respectively. In the fetus, serum concentrations of phosphate, PTHrP, and calcitonin are higher than in the pregnant woman, while fetal levels of 1,25-dihydroxyvitamin D 3 (calcitriol) and PTH are low and those of 25-hydroxyvitamin D 3 (calcidiol) approximate maternal values. In the neonate, total calcium and Ca 2 + concentrations decline in the first 24 hours after birth to values approximating 8 to 9 mg/dL (2.0–2.75 mmol/L) and 4.4 to 5.4 mg/dL (1.1–1.35 mmol/L), respectively; calcium levels then plateau and subsequently increase to the midnormal range by the third day of life. Neonatal concentrations of PTHrP and calcitonin decline after delivery, whereas values of PTH and calcitriol increase over the first 2 postnatal days. Subsequently, in healthy infants, Ca 2 + concentrations vary by postnatal age: 1 month—5.2 to 6.1 mg/dL = 1.28 to 1.52 mmol/L; 3 months—5.2 to 6.0 mg/dL = 1.30 to 1.49 mmol/L; 12 months—5.0 to 5.6 mg/dL = 1.24 to 1.39 mmol/L.
Clinical manifestations of hypocalcemia occurring in the neonate (defined as values of total calcium < 7.5–8.0 mg/dL and/or Ca 2 + < 4.4 mg/dL [1.1 mmol/L] in newborns with birth weights > 1500 g and < 7.0 mg/dL and/or Ca 2 + < 3.6 ng/dL [0.9 mmol/L] in newborns with birth weights < 1500 g) are principally those of neuromuscular hyperexcitability: irritability, hyperacusis, jitteriness, tremulousness, facial spasms, tetany, laryngospasm, and focal or generalized seizures. Nonspecific symptoms, such as apnea, tachycardia, cyanosis, emesis, and feeding problems may also occur. Causes of neonatal hypocalcemia may be considered in relation to the age of onset (before or after 72 hours of life = early/late) ( Tables 20.2A, 20.2B ).
|
|
| Gene Protein Chromosome OMIM | Disorder OMIM | Clinical/Biochemical Manifestations | Gene Function/Transmission |
|---|---|---|---|
| Autosomal dominant hypoparathyroidism | |||
| CASR Calcium sensing receptor 3q13.3-q21.1 601199 | Autosomal dominant hypocalcemia type 1, (isolated hypoparathyroidism) 601198 | Hypocalcemia-mediated increased neuromuscular irritability: paresthesias, tetany, seizures; may also result in hypokalemia & secondary hyperaldosteronism | Encodes calcium sensing receptor expressed on plasma membrane of parathyroid gland & renal tubules; gain-of-function variants increase sensitivity & response of CaSR to low serum concentrations of calcium, AD (may be associated with Bartter syndrome type 5 caused by coimpairment of renal tubular reabsorption of sodium chloride, AD 601198) |
| GNA11 Guanine nucleotide-binding protein, alpha-11 19p13.3 139313 | Autosomal dominant hypocalcemia type 2 (isolated hypoparathyroidism) 615361 | Hypocalcemia-mediated increased neuromuscular irritability: paresthesias, tetany, seizures | Encodes G-protein alpha subunit (Gα11) that initiates intracellular signal transduction after binding of Ca 2 + to CaSR, AD |
| Familial isolated hypoparathyroidism | |||
| PTH Parathyroid hormone 11p15.3 168450 | Familial isolated hypoparathyroidism 146200 | Hypocalcemia-mediated increased neuromuscular irritability: paresthesias, tetany, seizures | Encodes parathyroid hormone, AD/AR |
| GCM2 Glial cells missing, drosophila, homolog of, 2 6p24.2 603716 | Familial isolated hypoparathyroidism 146200 | Hypocalcemia-mediated increased neuromuscular irritability: paresthesias, tetany, seizures | Encodes transcription factor essential for differentiation of the parathyroid glands, AD/AR |
| FHL1 Four-and-a-half Lim domains 1 300163 Xq26.3 | Isolated hypoparathyroidism 146200 | Hypocalcemic seizures | Encodes gene essential for differentiation of the parathyroid glands, X-linked |
| Complex hypoparathyroidism | |||
| TBX1 T-box 1 22q11.21 602054 | DiGeorge syndrome (DGS) type 1 (deletion chromosome 22q11 syndrome) 188400 | Hypoparathyroidism, thymic hypoplasia, congenital heart anomalies, cleft palate, impaired renal function, dysmorphic facial features | T-box transcription factor embryonically expressed in the tissues adversely impacted in DGS; AD |
| NEBL Nebulette 10p12.31 605491 | DiGeorge syndrome type (complex) 2, 601362 | Thymic aplasia, congenital heart anomalies, cleft palate, impaired renal function, dysmorphic facial features | Protein expressed in cardiac & striated muscle, associated with actin, myofibrils, & cellular adhesion complexes, AD |
| CHD7 Chromodomain helicase DNA-binding protein 7 8q12.2 608092 | CHARGE syndrome 214800 | CHARGE = Coloboma, Heart anomaly, Choanal atresia, Retardation, Genital, & Ear anomalies | Regulator of neural crest gene expression & ribosomal RNA formation, AD |
| GATA3 GATA-binding protein 3 10p15 131320 | HDR (Barakat syndrome) 146255 | Hypoparathyroidism, sensorineural deafness, renal dysplasia | Transcription factor/ enhancer element required for development of parathyroid glands, auditory system, kidneys & for expression of genes encoding T-cell receptor subunits, AD |
| TBCE Tubulin-specific chaperone E 1q42.3 604934 | HRD (Sanjad-Sakati syndrome—SSS), 244460; Kenny-Caffey syndrome type 1 (KCS1) 241410 | HRD: Hypoparathyroidism, retardation, dysmorphism; KCS1: aforementioned + osteosclerosis & recurrent infections | Chaperone protein necessary for correct folding of tubulin subunits & stability of cellular cytoskeleton, AR |
| FAM111A Family with sequence similarity 111, Member A 11q12.1 615292 | Kenny-Caffey syndrome type 2 (KCS2), 127000; Gracile bone dysplasia (GBD), 602381 | KCS2: Hypoparathyroidism, retardation, dysmorphism; GBD: Hypocalcemia, thin but dense & fragile bones, may be lethal perinatally | Functional effect(s) in man unknown, (host range restriction factor in viruses), AD |
| Mitochondropathic hypoparathyroidism | |||
| Deletion mitochondrial chromosome | Kearns-Sayre syndrome (KSS) 530000 | Hypoparathyroidism, ophthalmoplegia, retinitis pigmentosa, sensorineural deafness, cerebellar ataxia, abnormal cardiac conductivity, myopathy, growth retardation, renal tubular dysfunction, hypoadrenocorticism, hypogonadism, diabetes mellitus | Mitochondrial genes encoding energy-generating electron transport proteins, Maternal, AD |
| Deletion mitochondrial chromosome | Mitochondrial encephalomyopathy, lactic acidosis, stroke-like episodes (MELAS) 540000 | Hypoparathyroidism, myopathy, ophthalmoplegia, neuropathy, cardiomyopathy, impaired cognition | Mitochondrial genes encoding energy-generating electron transport proteins, Maternal, AD |
| HADHB Hydroxylacyl-CoA dehydrogenase/3-Ketoacyl-CoA thiolase/Enoyl-CoA hydratase, beta subunit 2p23.3 143450 | Mitochondrial trifunctional protein deficiency syndrome, 609015 | Because of inability to metabolize an energy source, clinical presentation may vary from acute & lethal in the perinatal period to hepatic Reye-like syndrome in older infants to skeletal myopathy in adolescents | Nuclear gene encoding beta subunit of mitochondrial trifunctional protein catalyzing beta oxidation of long chain fatty acids, AR |
| Autoimmune hypoparathyroidism | |||
| AIRE Autoimmune regulator 21q21.3 607358 | Autoimmune polyendocrinopathy syndrome, type I 240300 | Hypoparathyroidism, hypoadrenocorticism, mucocutaneous candidiasis, alopecia, pernicious anemia, hypogonadism | Expressed in thymus, essential for recognition of self-antigens, AD/dominant negative/AR |
| Pseudohypoparathyroidism | |||
| GNAS GNAS complex locus 20q13.32 139320 | Pseudohypoparathyroidism type 1A, 103580 | Hypocalcemia, hyperphosphatemia, subnormal renal tubular cyclic AMP response to exogenous PTH, Albright hereditary osteodystrophy phenotype (AHO) | Inactivating mutations or deletions of maternal GNAS or biallelic paternal expression of GNAS (isochromosomes) lead to proximal renal tubular & skeletal resistance to PTH, AD |
| GNAS GNAS complex locus 20q13.32 139320 | Pseudohypoparathyroidism type 1B, 603233 | Hypocalcemia, hyperphosphatemia, subnormal renal tubular cyclic AMP response to exogenous PTH, normal erythrocyte cyclic AMP activity, normal phenotype | Maternally transmitted epigenetic methylation defect of maternal GNAS leads to its silencing & proximal renal tubular resistance to PTH, AD |
| GNASAS1 GNAS complex locus, antisense transcript 1 20q13.32 610540 | Pseudohypoparathyroidism type 1B 603233 | Hypocalcemia, hyperphosphatemia, absent renal tubular cAMP response to exogenous PTH, normal phenotype | Maternally transmitted deletion leads to paternal expression of GNASAS1 resulting in silencing of maternal GNAS because of an epigenetic methylation defect of & proximal renal tubular resistance to PTH, AD |
| STX16 Syntaxin 16 20q13.32 603666 | Pseudohypoparathyroidism type 1B 603233 | Hypocalcemia, hyperphosphatemia, absent renal cyclic AMP response to exogenous PTH, normal phenotype | Maternally transmitted epigenetic methylation defect of maternal GNAS leads to its silencing & proximal renal tubular resistance to PTH, AD |
| GNAS GNAS complex locus 20q13.32 139320 | Pseudohypoparathyroidism type 1C | Hypocalcemia, hyperphosphatemia, absent renal cyclic AMP response to exogenous PTH, AHO phenotype, normal erythrocyte cyclic AMP activity | Inactivating mutations or deletions in exon 13 of maternal GNAS lead to proximal renal tubular & skeletal resistance to PTH, AD |
| ? | Pseudohypoparathyroidism type 2, 203330 | Hypocalcemia, hyperphosphatemia, partial resistance to PTH, normal phenotype | Despite normal increase in urinary cyclic AMP after PTH administration, patients are resistant to phosphaturic effect of PTH |
| GNAS GNAS complex locus 20q13.32 139320 | Pseudopseudohypoparathyroidism, 612463 | Phenotype of AHO but normocalcemia | Skeletal resistance to PTH because of loss-of-function variants of paternal GNAS |
| PRKAR1A Protein kinase, cAMP dependent, regulatory, type 1 alpha 17q24.2 188830 | Acrodysostosis 1 101800 | Skeletal dysplasia: short stature, brachydactyly, facial dysostosis, nasal hypoplasia; with/without hormone resistance | Encodes a regulatory subunit of cyclic AMP-dependent protein kinase A requisite for intracellular signal transduction |
| PDE4D Phosphodiesterase 4D, cAMP-specific 5q11.2-q21.1 600129 | Acrodysostosis 2 614613 | Vide supra , developmentally challenged | Encodes enzyme that degrades cyclic AMP, thereby inhibiting intracellular signal transduction |
Early Neonatal Hypocalcemia
In the absence of hypoproteinemia, hypocalcemia occurring within the first 72 hours after birth is considered “early neonatal hypocalcemia.” It occurs most commonly in prematurely delivered or small-for-gestational-age, low birth weight (LBW), or asphyxiated neonates, or in those born to women with gestational or permanent forms of diabetes mellitus (rarely unsuspected maternal hyperparathyroidism) and is the consequence of subnormal PTH secretion in response to declining serum calcium values and delayed renal tubular phosphaturic response to PTH characteristic of the neonate, unusually prolonged secretion of calcitonin, and/or hypomagnesemia. Total calcium and Ca 2 + concentrations decline more rapidly from high intrauterine values to lower nadir levels in preterm than in term neonates. In LBW neonates, hypocalcemia may be further attributed to the rapid accretion of skeletal calcium in the presence of relative resistance to the calcium absorptive and reabsorptive effects of calcitriol on the intestinal tract and bone, respectively. Offspring of severely vitamin D–deficient mothers may become hypocalcemic shortly after birth. Hypocalcemia develops in approximately one-third of asphyxiated newborns who are products of complicated and compromised deliveries, including those associated with maternal diabetes mellitus, toxemia of pregnancy, vitamin D deficiency, use of anticonvulsant drugs, and (unsuspected) maternal hyperparathyroidism. In these infants, increased phosphate load caused by cellular injury, reduced calcium intake, and hypercalcitonemia are important pathogenetic factors in the development of hypocalcemia. Neonates born prematurely and those with intrauterine growth restriction, asphyxia, infections, respiratory distress syndrome, or other critical illness are often hypocalcemic.
Some 50% of infants of mothers with diabetes mellitus develop early neonatal hypocalcemia; the incidence may be reduced by strict maternal glycemic control. Its causes are multifactorial and include reduced placental transfer of calcium because of substantial maternal urinary excretion of calcium and magnesium, decreased neonatal secretion of PTH, hypercalcitonemia, hypomagnesemia (occurring in 40% of offspring of diabetic women), and limited intake and impaired absorption of ingested calcium. Maternal hypercalcemia caused by unsuspected hyperparathyroidism leads to increased transfer of calcium to the fetus and still further increase in in-utero serum calcium concentrations that suppress fetal PTH synthesis and release and stimulate calcitonin secretion—aberrations in homeostatic mechanisms that persist postpartum and may result in hypocalcemic tetany/seizures in her offspring. Suppression of PTH secretion may persist for several months and be undetected until symptomatic hypocalcemia develops after weaning of the infant from breast milk to higher phosphate containing cow milk formula. Maternal ingestion of large quantities of calcium carbonate in antacids has also led to neonatal hypocalcemia.
Hypocalcemia may occur in neonates with hyperbilirubinemia undergoing exchange transfusion and in those exposed to phototherapy. Neonates with acute rotavirus infection and severe diarrhea may present with hypocalcemic seizures. Aminoglycoside antibiotics (e.g., gentamycin) increase urinary excretion of calcium and magnesium, thereby facilitating the development of neonatal hypocalcemia. Compounds that complex with and sequester calcium, such as citrate (present in transfused blood), phosphates (that alter the calcium x phosphate product), and fatty acids (given as caloric supplements) lower Ca 2 + levels. Bicarbonate administered to correct acidosis increases calcium binding to albumin and thus lowers Ca 2 + values. Hypomagnesemia impairs release of PTH from the parathyroid glands. Hypocalcemia may also occur in hyperventilated infants with severe respiratory alkalosis, as well as in those with other causes of metabolic alkalosis. Phytates in soy milk bind calcium and phosphate and interfere with their absorption. Neonates and infants with malignant osteopetrosis type II and impaired osteoclastogenesis may present with either early or late neonatal hypocalcemia.
Late Neonatal Hypocalcemia
Late transient hypocalcemia (first developing when the neonate is > 72 hours of postnatal age) may be caused by increased intake of phosphate, hypomagnesemia, hypoparathyroidism, or vitamin D deficiency (see Tables 20.2A, 20.2B ). Neonatal hypocalcemia may develop after 3 days of age in offspring born in the late winter-early spring of the year to multiparous women with inadequate intake of vitamin D or exposure to sunlight. High phosphate content of evaporated milk or modified cow milk formulas may lead to formation of poorly soluble calcium salts limiting the intestinal absorption of calcium while raising serum phosphate values. Premature introduction of fiber-containing cereals into the infant’s diet also decreases calcium absorption. Affected infants may have an associated defect in renal phosphate excretion or coexisting vitamin D deficiency. Hyperphosphatemia and hypocalcemia may initially suggest hypoparathyroidism, but serum PTH concentrations are usually normal or modestly elevated in infants with excessive phosphate loading in response to reciprocal reduction in serum calcium; markedly elevated or persistently high PTH values raise the question of whether pseudohypoparathyroidism (PHP, rarely acrodystostosis—a skeletal dysplasia with hormone resistance related to inactivating variants of PRKAR1A or PDE4D , vide infra) may be present. Newborns and infants with chronic renal insufficiency because of renal hypoplasia or obstructive nephropathies often are hypocalcemic and hyperphosphatemic with elevated serum PTH levels as well, but they are also azotemic. Hypomagnesemia leads to impaired secretion of PTH and decreased peripheral responsiveness to PTH and may be transient or related to congenital abnormalities of intestinal absorption or renal tubular reabsorption of magnesium. Hypermagnesemia may occasionally be associated with neonatal hypocalcemia.
Hypocalcemia and hypophosphatemia caused by fetal/neonatal deficiency of vitamin D occurs in offspring of mothers with substantial deficiency of vitamin D (either for cultural or socioeconomic reasons), impaired hepatic 25-hydroxylation of cholecalciferol or renal 25-hydroxyvitamin D-1α hydroxylase activity or loss-of-function mutations of the vitamin D receptor (VDR). Hypovitaminosis D may develop in an older breastfed infant of a vegetarian mother who shields herself from sunlight and ingests a diet low in vitamin D. Marginal deficiency of vitamin D in neonates and infants is much more common than has been recognized heretofore. “Late-late” neonatal hypocalcemia occurs in premature infants with low bone mass at 3 to 4 months of age in whom the intake of calcium, phosphate, and vitamin D has been marginal; it is perhaps caused by avid deposition of available calcium into bone. Hypocalcemia caused by vitamin D deficiency may develop rather acutely and in the absence of clinical or radiographic signs of rickets in the older infant and young child ingesting an elimination diet low in vitamin D because of severe allergies and/or maintained indoors with limited exposure to sunlight.
Hypocalcemia initially manifesting after 72 hours of age often heralds the presence of significant compromise of calcium homeostatic mechanisms, such as those associated with hypoparathyroidism because of malformation of the parathyroid glands (e.g., the DiGeorge syndrome or variant of a gene critical for embryogenesis of these structures) or functional error in PTH secretion (e.g., an abnormality in the activity of the calcium sensing receptor [CaSR]).
Hypoparathyroidism
Hypoparathyroidism presenting in infancy is often transient and related to delayed developmental maturation of parathyroid gland function; it may resolve within the first several weeks of life (see Tables 20.2A, 20.2B and Fig. 20.1 ). When persistent, hypoparathyroidism may be caused by an error in the embryogenesis of the parathyroid glands or in the synthesis or secretion of PTH or to peripheral unresponsiveness to PTH (functional hypoparathyroidism, i.e., PHP, occurs in patients who are resistant to PTH [vide infra]). Familial isolated congenital hypoparathyroidism may be transmitted as an autosomal dominant, autosomal recessive, or X-linked recessive trait caused by loss-of-function mutations in genes required for differentiation of the parathyroid glands leading to congenital aplasia or hypoplasia of these structures occurring as a unitary disorder. Thus, familial isolated hypoparathyroidism (OMIM 146200) has been related to inactivating mutations in PTH , glial cells missing ( GCM2 ), and four-and-a-half Lim domains 1 ( FHL1 ) and autosomal dominant hypocalcemia with suppressed secretion of PTH because of gain-of-function variants of CASR and guanine nucleotide binding protein alpha 11 ( GNA11 ). Inactivating mutations in the exons encoding the signal peptide of PTH interfere with the processing of preproPTH to the bioactive 84 aa functional PTH molecule resulting in hypoparathyroidism that may be transmitted as an autosomal dominant or recessive characteristic. Depending on the specificity of the immunoassay for PTH, serum levels of PTH may be low, normal, or even high in these patients. GCM2 is a gene with five exons that encodes a 506 aa deoxyribonucleic acid (DNA)-binding transcription factor whose expression is restricted to the parathyroid glands. Intragenic deletions or homozygous missense inactivating mutations in exons 2, 3, and 5 of GCM2 result in hypoparathyroidism in humans. Mutations in GCM2 exons 2 and 3 (encoding DNA binding and transactivation domain 1) lead to impaired protein synthesis and stability and autosomal recessive transmission of congenital hypoparathyroidism whereas those in exon 5 (encoding transactivation domain 2) lead to mutations with a dominant negative effect and autosomal dominant transmission of this disorder. (Expression of GCM2 takes place soon after specification of parathyroid cells and is dependent on normal transcriptional function of GATA3 , the gene mutated in patients with the Barakat syndrome of hypoparathyroidism-deafness-renal dysplasia [HDR] [vide infra]. Activating mutations of GCM2 are associated with hyperparathyroidism [vide infra].) X-linked hypoparathyroidism is associated with agenesis of the parathyroid glands; the disorder is caused by loss-of-function variants of FHL1 (encoding Four-and-a-half Lim domains 1, OMIM 300163), a gene whose product is essential for differentiation of the parathyroid glands. FHL1 is a 280-amino-acid (aa) protein with a LIM domain (double zinc finger motif) that is also expressed in testes, cardiac, and skeletal muscle.
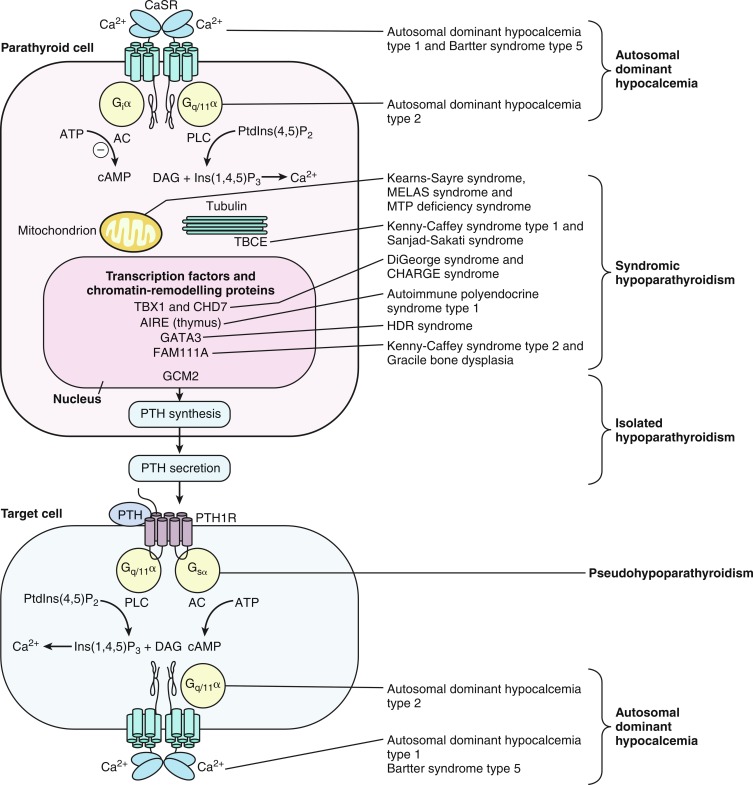
Hypocalcemia associated with impaired secretion of PTH caused by monoallelic activating mutations in CASR have been identified in neonates, infants, children, and adults. Hypercalciuric hypocalcemia is an autosomal dominantly transmitted form of hypoparathyroidism that is caused by gain-of-function mutations in CASR (autosomal dominant hypocalcemia type 1) or GNA11 (autosomal dominant hypocalcemia type 2) that result in enhanced “sensitivity” of the CaSR to the PTH-suppressive effects of Ca 2 + in the chief cells of the parathyroid glands and in renal cells in the thick ascending limb of the loop of Henle (TALH). The CaSR monitors plasma concentrations of Ca 2 + , whereas GNA11 is the alpha subunit—guanine nucleotide binding protein of the trisubunit guanosine protein binding receptor (GPCR) transmitting the signal of the CaSR to intracellular signal transduction pathways. A lowered “set-point” of the CaSR or enhancement of the postreceptor signal transduction system itself enables PTH secretion to be suppressed and renal tubular reabsorption of calcium to be depressed (thus increasing urine calcium excretion) by extremely low concentrations of Ca 2 + resulting in marked hypercalciuria. Activating mutations (e.g., p.Lys47Asn, p.Leu616Val, p.Phe788Leu) may be scattered throughout CASR but occur predominantly in the second peptide loop of its extracellular domain. Some activating monoallelic variants (e.g., p.Cys141Trp, p.Leu125Pro, p.Ala843Glu) of CASR may also inhibit function of the renal outer medullary potassium channel (encoded by KCNJ1 , OMIM 600359), leading to a Bartter-like syndrome with hypokalemic metabolic alkalosis, hyperreninemia, hyperaldosteronism, and hypomagnesemia, as well as hypercalciuric hypocalcemia (designated Bartter syndrome type 5, OMIM 601198). These paired metabolic defects are partially responsive to treatment with hydrochlorothiazide and low doses of calcitriol. However, children with hypercalciuric hypocalcemia caused by gain-of-function mutations in CASR are very sensitive to even low doses of calcitriol that can lead to even more marked hypercalciuria and to nephrocalcinosis. Thus management of these patients has been difficult. Administration of recombinant human PTH 1-34 to a 14-month-old hypocalcemic male infant with a de novo nonsense mutation in CASR (p.Leu727Gln) for 17 months partially restored calcium homeostasis with increased but still subnormal serum levels of calcium, while urinary excretion of calcium decreased into the normal range. During treatment, the child was clinically asymptomatic, did not develop nephrocalcinosis, and tolerated the drug well. However, administration of PTH to such a patient may increase urine calcium excretion and the risk of nephrocalcinosis. Use of calcilytic agents (type II calcimimetics), such as cinacalcet that antagonize the effects of calcium upon the CaSR in the parathyroid glands and renal tubules may prove therapeutically useful in these patients. Neonates with variants of GNA11 may not have the same degree of hypercalciuria as do those with mutant CASR .
The most commonly encountered forms of hypoparathyroidism are those associated with constellations of congenital abnormalities (see Tables 20.2A, 20.2B and see Fig. 20.1 ). The most frequent complex form of hypoparathyroidism is that associated with the DiGeorge syndrome type 1 (OMIM 188400), a disorder that occurs with a frequency of 1:4000 births and is present in approximately 70% of children with isolated hypoparathyroidism. In many infants with the DiGeorge syndrome, hypoparathyroidism partially remits over time only to reappear in periods of stress, such as infections or trauma. The DiGeorge syndrome is a neurocristopathy—the result of disturbed migration of cervical neural crest cells and consequent maldevelopment of tissues of neural crest origin derived from the third and fourth pharyngeal pouches and first to fifth branchial arches. DiGeorge syndrome type 1 is associated with microdeletions of chromosome region 22qll.2 (del22q11.2—the DiGeorge critical region), a segment on which more than 35 genes are sited and thus is a contiguous gene syndrome (a disorder caused by deletion of several adjacent genes that when individually mutated may result in a distinctive clinical feature but when collectively lost leads to a group of apparently unrelated clinical findings). The chromosome 22q11.2 microdeletion is contained within regions of low copy number repeats, and it is this characteristic that results in unequal segmental exchange between the paired 22 nd chromosomes during meiosis. Subjects with the DiGeorge syndrome usually have the triad of: (1) hypocalcemia because of hypoplasia of the parathyroid glands often manifest in the neonatal period but which may not be detected until an older age, (2) defective T lymphocyte function and impaired cell-mediated immunity because of partial or complete absence of thymic differentiation leading to increased frequency of viral and fungal infections and propensity of autoimmune disorders, and (3) conotruncal defects of the heart or aortic arch (Tetralogy of Fallot, ventricular septal defect, interrupted or right aortic arch, truncus arteriosus, vascular ring). To a substantial extent, DiGeorge syndrome type 1 is related to loss or deleterious variants of T-box 1 ( TBX1 , OMIM 602054) within the 22q11.2 chromosomal segment. Experimental disruption of Tbx1 impairs development of the pharyngeal arch arterial vasculature, whereas introduction of null mutations in Tbx1 results in anomalies of the cardiac outflow track and hypoplasia of the thymus and parathyroid glands. The transcription factor encoded by TBX1 is part of a network of gene products (including those encoded by ISL1, SHH, FOXA2, FOXC2 ) that controls development of the parathyroid glands and thymus by regulating expression of GATA3, GCM2 , and PAX9. Mutations of TBX1 specifically account for five major manifestations of the DiGeorge syndrome: parathyroid hypoplasia and hypocalcemia, thymic aplasia, cardiac anomalies, unusual facial features (low set ears, micrognathia, slanted eyes, short palpebral fissures and philtrum, small mouth), and cleft palate with velopharyngeal insufficiency. TBX1 haploinsufficiency can also cause isolated hypoparathyroidism. Also within this two megabase microdeletion at chromosome 22q11.2 is HIRA (histone cell cycle regulation, OMIM 600237), a transcription regulatory factor that is expressed in developing heart and upper body neural crest elements and is necessary for normal cardiac development. Another critical gene sited at chromosome 22q11.2 is UFD1L (ubiquitin fusion degradation 1-like, OMIM 601754), whose product is important for the posttranslational processing of proteins and/or their degradation by interaction with the ubiquitin fusion protein. Experimentally, the DiGeorge syndrome has been linked to genes encoding endothelin-1, vascular endothelial growth factor, and fibroblast growth factor-8 ( Fgf8 , a target gene for TBX1 ). In the mouse hypomorphic for Fgf8, there are cardiovascular, craniofacial, parathyroid, and thymic defects—an experimental phenocopy of the human del22q11.2 syndrome. (Maternal diabetes mellitus, alcoholism, or ingestion of retinoic acid may occasionally be associated with the DiGeorge syndrome in the offspring.)
Prenatally, the presence of the DiGeorge syndrome may be considered when fetal ultrasonography reveals an interrupted aortic arch or truncus arteriosus and may be confirmed by appropriate studies (microarray, fluorescent in situ hybridization [FISH]) on samples of chorionic villi or amniotic fluid. Other clinical features of the DiGeorge syndrome include: growth retardation, renal dysplasia, gastrointestinal malformations (esophageal atresia, anal atresia), cervical spine instability, impaired vision, and ocular malformations, malformation of the cerebral cortex (perisylvian polymicrogyria), and developmental delay. DiGeorge syndrome type 1 (OMIM 188400) may occur sporadically or be transmitted as an autosomal dominant trait. DiGeorge syndrome type 2/Velocardiofacial syndrome complex 2 (OMIM 601362) has been associated with interstitial deletion of chromosome 10p13 but attributed, in part, to loss of NEBL (Nebulette, OMIM 605491), sited at chromosome 10p12.31. NEBL encodes a protein expressed in cardiac and striated muscle that associates with actin, myofibrils, and cellular adhesion complexes; DiGeorge syndrome type 2 is transmitted as an autosomal dominant characteristic. The DiGeorge syndrome has also been associated with microdeletions of chromosomes 18q21.33 and 4q21.2-q25—indicative of the cascade of genes likely involved in the generation of this phenotype. In addition to DiGeorge syndrome type 1, deletion of chromosome 22q11.2 has been associated with the velocardiofacial and other syndromes. Collectively, these syndromes display similar facial features (ocular hypertelorism, lateral displacement of inner canthi, short palpebral fissures, swollen eyelids, dysmorphic “segmented” nose, small mouth, low set ears with abnormally folded pinnae, short philtrum, micrognathia, malar hypoplasia, velopharyngeal insufficiency with/without cleft palate), olfactory dysfunction, short stature, nonverbal learning disabilities, and various psychological maladies. Takao velocardiofacial syndrome (included in OMIM 188440) consists primarily of the typical cardiac defects described earlier that may also be associated with hypocalcemia; Shprintzen velocardiofacial syndrome (OMIM 192430) is characterized by craniofacial and palatal defects and cardiac anomalies; Cayler cardiofacial syndrome (OMIM 125520) is associated with partial unilateral facial paresis because of hypoplasia of the depressor angulioris muscle and anomalies of the heart and aorta. These syndromes have been grouped as the CATCH-22 syndromes of cardiac defects, abnormal face, thymic hypoplasia, cleft palate, hypocalcemia.
Hypocalcemia has been observed in some subjects with microduplication of chromosome 22q11.2, a copy number variant; clinical characteristics of individuals with this genetic anomaly vary from those who are entirely normal to patients with multiple congenital anomalies, severe developmental delay, autism, and schizophrenia. The duplication chromosome 22q11.2 syndrome appears to be transmitted as an autosomal dominant characteristic whose expression is modified by other factors. The pathophysiology of hypocalcemia in affected subjects is uncertain. In one family in which the proband had the DiGeorge syndrome associated with del22q11.2, the normal father had the same anomaly on one of his 22 nd chromosomes and dup22q11.2 on his other 22 nd chromosome; paternal quantitative expression of the genes located on chromosome 22q11.2 was normal indicating that the adverse effects of the 22q11.2 deletion were compensated by the 22q11.2 duplication.
There are several other syndromes with multisystem involvement and hypoparathyroidism. Hypoplasia of the parathyroid glands and consequent hypoparathyroidism may be observed in patients with the CHARGE syndrome (coloboma, heart anomaly, choanal atresia, retardation, genital and ear anomalies, OMIM 214800) as a result of deletion of chromosome 8q12 or specifically to a heterozygous inactivating variant of CHD7 (chromodomain helicase DNA-binding protein 7, OMIM 608092). CHD7 encodes a transcription regulating factor essential for differentiation of the neural crest that may coassociate with TBX1 . CHD7 is an adenosine triphosphate (ATP)-dependent chromatin remodeler that regulates movement of nucleosomes. The incidence of the CHARGE syndrome is approximately 1/10,000 births. The CHARGE and DiGeorge syndromes share several anomalies including hypoparathyroidism, cardiac and renal anomalies, cleft palate, ear abnormalities, and developmental delay. Indeed, hypoparathyroidism is more common (72%) in neonates with CHARGE than in those with the DiGeorge syndrome (26%).
The Barakat or HDR syndrome of hypoparathyroidism, sensorineural deafness, and renal disease (dysplasia, steroid-resistant nephrosis with progressive renal failure—OMIM 146255) has been attributed to monoallelic deleterious variants of GATA3 encoding GATA-binding protein-3 (OMIM 131320), a zinc-finger transcription factor/enhancer element that is required for development of the parathyroid glands, auditory system, and kidneys and for expression of genes encoding the four T-cell receptor subunits. Heterozygous inactivating mutations of GATA3 primarily involving loss of its carboxyl terminal DNA-binding segment are transmitted as an autosomal dominant disorder. Insertions, missense, and nonsense mutations in GATA3 have also been identified in patients and families with HDR. GATA3 is a zinc-finger transcription factor that regulates expression of GCM2 and thus is critical for the embryonic development of the parathyroid glands, as well as for the kidneys, otic vesicles, and thymus. The parathyroid glands of these children are hypoplastic or absent. Hypocalcemia may be present in the newborn period or unrecognized until later childhood. Malformations of the uterus and vagina (didelphic uterus, septate vagina) may be present in females with this disorder.
The Sanjad-Sakati syndrome of congenital hypoparathyroidism, mental retardation, and facial dysmorphism (depressed nasal bridge, long philtrum, thin upper lip, long filtrum) (HRD, OMIM 241410) is caused by biallelic loss-of-function mutations (often a 12 base pair deletion in the third coding exon) in TBCE (tubulin-specific chaperone E, OMIM 604934). TBCE is a chaperone protein that is essential for formation/folding and stability of microtubules—cytosolic structures composed of heterodimeric α- and β-tubulin subunits that form the cytoskeleton, mitotic apparatus, cilia, and other cellular components; this chaperonin assists in the correct folding of α- and β-tubulin subunits and the formation of α-β-tubulin heterodimers. The α- and β-tubulin subunits and TBCE are necessary for normal embryogenesis of the parathyroid glands. Mutations in TBCE result in lowered microtubule formation and consequently in decrease in subcellular components, such as the cytoskeleton, Golgi apparatus, and endosomal compartments required for normal intracellular movement of proteins, as well as formation of the mitotic apparatus and cilia. The majority of infants with HRD sustain intrauterine growth retardation and manifest hypocalcemic seizures with low serum concentrations of PTH and normal phosphaturic responses to exogenous PTH in the first several weeks and months after birth. Children with HRD are short, developmentally delayed, and seizure prone; they have medullary stenosis of the long bones and other skeletal anomalies; they are microcephalic with faces characterized by deeply recessed eyes or microphthalmia, depressed nasal bridge, beaked nose, long philtrum, thin upper vermillion border, micrognathia, and large floppy earlobes. The cardiovascular system of these patients is intact, but as infants, they are susceptible to life-threatening pneumococcal infections. Additional features in HRD subjects as assessed by neuroimaging include adenohypophyseal hypoplasia and attenuation of the pituitary stalk, infundibulum, and corpus callosum.
When a patient with the Sanjad-Sakati syndrome—HRD triad also manifests cortical thickness and medullary stenosis of the long bones, osteosclerosis of the skull, and susceptibility to recurrent infections and has biallelic variants of TBCE , the complex is termed the Kenny-Caffey syndrome type 1 (KCS1, OMIM 244460). Neonates with KCS1 are often severely hypocalcemic early in the postnatal period. As children they are short, with microcephaly and craniofacial anomalies because of absence of diploic space in the skull, osteosclerosis, and thickening of the cortices of the long bones with narrowing of the medullary compartment, normal or mildly delayed development, and increased susceptibility to recurrent bacterial infections. Interestingly, the identical mutation in TBCE —a homozygous 12-bp deletion in exon 2—may result in either the HRD or KCS1 phenotype in a specific family. The clinical, laboratory, and radiographic findings in patients with Kenny-Caffey syndrome 2 (KCS2, OMIM 127000) are similar to those in subjects with KCS1, except that patients with KCS2 have normal intelligence. KCS2 is the result of monoallelic loss-of-function mutations in FAM111A (family with sequence similarity 111, Member A, OMIM 615292). Allelic to KCS2 is gracile bone dysplasia (OMIM 602361), in which the bones are thin, slender, and brittle, the diaphyses are dense, the basal cranial sutures close prematurely, there is microphthalmia, and hypocalcemia because hypoparathyroidism is common. Hypomagnesemia is often present as well. Heterozygous variants in FAM111A have been identified in these patients. FAMIIIA encodes a factor whose expression is lowest in stage G0 of the cell cycle but whose primary function is unknown; it has been suggested that FAMIIIA may interact with TBCE to regulate gene expression or function.
Mitochondria are cytoplasmic organelles that are primarily donated to the embryo within the cytoplasm of the fertilized ovum and are therefore of maternal origin. Mitochondria are the primary intracellular site of respiration and energy utilization. The mitochondrial genome consists of a single circular chromosome with 37 embedded genes. Thirteen mitochondrial genes encode proteins required for electron transport and energy generation; 22 mitochondrial genes encode transfer ribonucleic acids (RNAs) required for protein synthesis. In addition, there are mitochondrial proteins whose genes reside in the cell’s nucleus but that are expressed within mitochondria. Thus mitochondrial dysfunction leading to hypoparathyroidism may be caused by deletion and/or duplication of mitochondrial DNA encoding genes intrinsic to the mitochondrion itself or to variants of nuclear DNA encoding a protein expressed within the mitochondrion. Clinical manifestations of mitochondrial DNA mutations almost always involve muscle dysfunction, as well as other tissues depending upon the postconceptual age and cellular site(s) in which the mitochondrial DNA variation occurs. The Kearns-Sayre syndrome (OMIM 530000) is associated with agenesis or dysgenesis of the parathyroid glands leading to hypoparathyroidism. Primary manifestations of this syndrome include progressive external ophthalmoplegia, pigmentary retinopathy, sensorineural deafness, cerebellar ataxia, abnormal cardiac conductivity, myopathy, growth retardation, and renal tubular dysfunction, as well as hypoadrenocorticism, hypogonadism, and diabetes mellitus. Both deletions and duplications of mitochondrial DNA may be found in patients with this syndrome. The syndrome of mitochondrial encephalomyopathy, lactic acidosis, and stroke-like episodes (MELAS, OMIM 540000) is also associated with hypoparathyroidism, as well as myopathy, ophthalmoplegia, neuropathy, cardiomyopathy, impaired cognition, and diabetes mellitus. Biallelic mutations in HADHB (Hydroxylacyl-CoA dehydrogenase/3-Ketoacyl-CoA thiolase/Enoyl-CoA hydratase, beta subunit, beta subunit, OMIM 143450) encoding mitochondrial trifunctional protein impair intramitochondrial beta oxidation of fatty acids resulting in the inability to use an energy source. Depending upon the extent of the enzymatic deficiency, clinical presentations may vary from acute and lethal in the perinatal period to a hepatic Reye-like syndrome in older infants to a skeletal myopathy in adolescents. Occasionally, hypoparathyroidism may occur in these patients.
PHP is a conglomerate of clinical disorders most often characterized by end-organ insensitivity to the biological effects of PTH (and PTHrP) resulting in hypocalcemia despite substantial secretion of endogenous PTH. The PTH receptor ( PTH1R , OMIM 168468) is a heptahelical transmembrane G-protein–coupled structure that after binding to extracellular PTH or PTHrP initiates intracellular signal transduction by changing its configuration, thereby enabling the linked submembrane stimulatory guanine nucleotide-binding protein (Gs-protein) to exchange guanosine triphosphate (GTP) for guanosine diphosphate (GDP) on the alpha subunit ( GNAS , OMIM 139320) of the heterotrimer of alpha, beta, and gamma subunits that together comprise the Gs-protein. After substitution of GTP for GDP on the α subunit, G sα dissociates from its linked βϐ subunits. G sα then stimulates intracellular adenylyl cyclase ( ADCY3 , OMIM 600291) enzymatic activity, thereby converting ATP to cyclic adenosine monophosphate (AMP) and releasing it from the intracellular surface of the plasma membrane of the PTH-responsive cell. Cyclic AMP in turn binds to the regulatory alpha subunit of protein kinase A or PKA ( PRKAR1A , OMIM 188830), thereby initiating intracellular signaling. The catalytic subunits of PKA then phosphorylate a number of intracellular proteins including the cyclic AMP- responsive binding protein ( CREB1 , OMIM 123810) that in turn initiate transcription of cyclic AMP target genes. CREB1 is inactivated by one of several phosphodiesterases encoded by PDE4D (OMIM 600129) and PDE3A . (OMIM 123805). After G sα has propagated signal transmission through activation of adenylyl cyclase, the intrinsic GTPase activity of G sα hydrolyzes the attached GTP to GDP, thereby halting further signal transduction.
Neonates with biallelic loss-of-function mutations in PTH1R are functionally hypoparathyroid despite elevated serum concentrations of PTH and thus represent a form of “PHP.” Because of subresponsiveness to PTHrP in utero, fetal bone formation is abnormal resulting in Blomstrand chondrodysplasia—an osteochondrodystrophy characterized by short extremities and advanced skeletal and dental maturation—abnormalities detectable in utero by fetal ultrasonography. Histologically, the proliferative zone of the cartilage growth plate is narrowed with relatively few resting and proliferating chondrocytes, whereas the hypertrophic zone is composed of irregular columns of chondrocytes. Transmitted as an autosomal recessive trait, its clinical characteristics include polyhydramnios, hydrops fetalis, short-limbed dwarfism, facial anomalies, aberrant tooth development, aplasia of the nipples and breasts, hypoplastic lungs, preductal aortic coarctation, and neonatal hypocalcemia and hyperphosphatemia despite elevated serum concentrations of PTH. Although Blomstrand osteochondrodysplasia (OMIM 215045) is usually lethal, skeletal malformations may be more (type I) or less severe (type II). Mutations in PTH1R that result in complete absence of normal protein (e.g., Arg104Ter) are designated type I while mutations that permit some PTH1R synthesis (Pro132Leu) result in type II Blomstrand osteochondrodysplasia. Eiken chondrodysplasia (OMIM 600002) is also caused by biallelic loss-of-function mutations in PTH1R but is clinically and radiographically distinct from Blomstrand osteochondrodysplasia as affected subjects have mild growth retardation, markedly delayed epiphyseal ossification, multiple epiphyseal dysplasia, and persistent islands of cartilage in the pelvis. Variants of PTH1R that occur in patients with the Eiken syndrome are located in the carboxyl terminal portion of the gene/protein (e.g., pArg485Ter). Heterozygous inactivating mutations of PTH1R may result in an autosomal dominant nonsyndromic failure of tooth eruption (OMIM 125350).
Classically, PHP is the term applied to the clinical state that is associated with abnormalities of the signaling pathway that transmits the message conveyed by interaction of PTH/PTHrP with PTH1R. Neonates and infants with PHP are often hypocalcemic and hyperphosphatemic with elevated serum levels of PTH, whereas older patients with PHP exhibit the characteristic phenotype and skeletal malformations of Albright hereditary osteodystrophy ([AHO]; growth retardation; brachydactyly of the third, fourth, and fifth metacarpal bones; round face; impaired dentinogenesis; and subcutaneous ossifications) with or without biochemical abnormalities. The AHO phenotype of growth retardation and brachydactyly is the result of accelerated closure of cartilaginous growth plates in long bones—the consequence of G sα haploinsufficiency in chondrocytes and early phase osteoblasts that accelerates osteoblast maturation. Palpable heterotopic subcutaneous ossifications, at times associated with a bluish discoloration of the overlying skin, develop in 70% of patients with AHO associated with PHP1a and pseudopseudohypoparathyroidism (PPHP); their number and size may increase over time and become acutely or chronically painful. Other unusual consequences of PHP1a include spinal stenosis, carpal tunnel syndrome, sensorineural and conductive hearing loss, olfactory impairment, sleep apnea, and asthma. Consistent with resistance to endogenous PTH, administration of exogenous PTH does not reduce the number of sodium-phosphate cotransporters (NaPi2a, NaPi2c) and thus does not increase renal excretion of phosphate or cyclic AMP.
GNAS is an imprinted gene; although GNAS is expressed from both maternal and paternal GNAS genes in many tissues, GNAS is expressed only from the maternal gene in the proximal renal tubule, thyroid, gonads, and adenohypophysis (vide infra). Thus in the proximal renal tubule GNAS expression is “imprinted”; that is, there is differential gene expression depending on the parent of origin of the allele. GNAS is composed of 13 exons; it has multiple transcripts that arise through splicing of four unique first exons onto shared exons 2 to 13 ( Fig. 20.2 ). Transcripts of GNAS include: (1) the G sα transcript—a protein that stimulates adenylyl cyclase and generates cyclic AMP—G sα is expressed by both maternal and paternal alleles in most tissues (including skin, white adipose tissue, chondrocytes, bone); however, only the maternal allele of GNAS is expressed in the proximal renal tubule, thyroid, gonads, and anterior pituitary (because of silencing of paternal GNAS caused by an epigenetic imprint) ; (2) XLαs yields a G sα isoform that is specifically expressed in neuroendocrine and nerve tissues and is identical to G sα except that it has a very long amino terminal sequence of amino acids; it is expressed only by the paternal allele; (3) the neuroendocrine secretory protein-55 (NESP55) transcript is a chromogranin-like protein that is expressed in neuroendocrine tissues but only by the maternal GNAS allele; and (4) the alternative first exon A/B (exon 1A) transcript is expressed ubiquitously but only at low levels and by the paternal GNAS allele and is not translated. The promoters of XLαs, NESP55, and exon 1A transcripts lie within the 5′ differentially methylated region (DMR) of GNAS . Methylation of the promoter usually silences expression of that GNAS transcript.
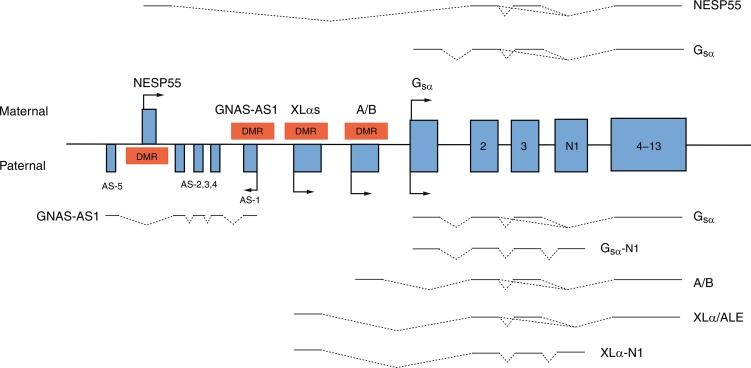
Loss-of-function mutations in GNAS or epigenetic (methylation) aberrations that result in failure of expression of a parental G sα transcript of GNAS lead to PHP types IA, IB, and IC and PPHP. PHP type 1A (PHP1a, OMIM 103580) is associated with resistance to protein hormones that signal through GPCRs and is the consequence of loss-of-function mutations in the maternal allele that encodes GNAS . Inactivating variants of maternal GNAS (functionally resulting in biallelic inactivity of GNAS and near total loss of G sα activity in the proximal renal tubule) lead to resistance to the biological effects of PTH in the proximal renal tubule and decreased reabsorption of filtered calcium resulting in hypocalcemia, exaggerated proximal renal tubular reabsorption of filtered phosphate leading to hyperphosphatemia, and elevated serum concentrations of PTH. In response to exogenous PTH, neither the urinary excretion of cyclic AMP nor that of phosphate increases. Erythrocyte G sα activity is subnormal in patients with PHP1a. (Resistance to thyroid-stimulating hormone [TSH] also occurs in patients with PHP1a. Thus PHP1a may be suspected in a neonate with hypocalcemia in whom hyperthyrotropinemia has been detected in the neonatal screening study for congenital hypothyroidism. ) More than 200 heterozygous loss-of-function mutations in maternal GNAS have been described. A four base pair deletion in exon 7 (codons 188-189) in GNAS that leads to a frameshift and premature stop codon has been found in a number of families with PHP1a and appears to be a “mutational hotspot” as it impairs DNA polymerization and replication. Other mutations alter intracellular movement of GNAS protein (p.Leu99Pro, p.Ser250Arg), increase the rate of release of GDP (p.Arg258Trp, p.Ala366Ser), or impair coupling of G-protein to PTH1R (p.Arg385His). Inasmuch as GNAS is expressed by both maternal and paternal alleles in the distal renal tubule where filtered calcium is also reabsorbed, nephrocalcinosis does not usually develop in patients with PHP1a.
Although patients with PHP type IB (PHP1b, OMIM 603233) generally have a normal phenotype (except for mild brachydactyly or obesity), they are hypocalcemic, hyperphosphatemic, and resistant to the biological effects of exogenous PTH and to TSH. PHP type IB occurs only in the offspring of obligate female carriers in whom loss of maternal GNAS expression in the kidney results in selective proximal renal tubular resistance to PTH; inasmuch as skeletal expression of both maternal and paternal GNAS is intact, bone formation is normal. PHP type IB is the result of defects in methylation of cytosine nucleotides within maternal GNAS resulting in its inactivation/conversion to a paternal GNAS epigenotype and the same functional consequences as those observed in patients with PHP type IA inasmuch as maternal GNAS is “silenced” in imprinted tissues, particularly the kidney. In some instances, this maternal epigenetic error has been transmitted by a maternal carrier to her offspring. In some patients, PHP type IB is caused by deletions within a DMR of GNAS or to loss of a 5′ cis-acting imprinting control center that is essential for methylation of the GNAS DMR that regulates expression of maternal GNAS in the proximal renal tubule. PHP type IB may also be caused by microdeletions of maternal GNAS exons AS3-4 or loss of methylation in maternal exons A/B of the differentially methylated region resulting in silencing of maternal GNAS or to variants of GNAS-AS1 or STX16. GNAS-AS1 (GNAS complex locus, antisense transcript 1, OMIM 610540) is embedded within the GNAS coding region, whereas STX16 (Syntaxin16, OMIM 603666) is positioned immediately centromeric (5′) to GNAS on the long (q) arm of chromosome 20; mutations (loss of exons 3-6) of maternal STX16 are associated with loss of methylation of GNAS exons A/B. Nevertheless, the genetic basis for most patients with sporadic PHP1b remains unknown; some of these patients appear to have global epigenetic methylation defects. Paternal uniparental isodisomy of the long arm of chromosome 20 (site of GNAS ) may be the cause of PHP1b in some patients. These patients have two normal paternal 20 th chromosomes but no functional GNAS in specific tissues (i.e., proximal renal tubules). Imprinting defects involving GNAS have also been identified in patients with AHO IB and microdeletions of chromosome 2q37. Inasmuch as bone is responsive to PTH in patients with AHO IA/IB, an occasional subject may develop osteitis fibrosa cystica over time. Nevertheless, because PTH also has anabolic effects on endocortical bone, bone mineralization in some patients with PHP1a/1b may be increased.
Patients with PHP type IC have the AHO phenotype and are hypocalcemic and hyperphosphatemic, but erythrocyte Gαs activity is normal. PHP type I is related to variants of the maternal GNAS allele in exon 13 (see Fig. 20.2 ) near its carboxyl terminal, often leaving intact the adenylate cyclase activity region. Patients with PHP type II (OMIM 203330) have hypocalcemia, hyperphosphatemia, and elevated serum levels of PTH but a normal phenotype. In response to exogenous PTH, patients with PHP type II increase the urinary excretion of cyclic AMP but do not increase urine phosphate excretion, indicating a defect in intracellular signaling distal to generation of adenylyl cyclase. Erythrocyte G sα activity is normal in patients with PHP type II. The pathogenesis of PHP type II has not as yet been identified, but in some subjects it has been suggested that it may be related to vitamin D deficiency and PTH resistance secondary thereto; others may be ingesting anticonvulsant medications that accelerate degradation of vitamin D and its bioactive metabolites. Subjects with PPHP (OMIM 612463) caused by inactivating mutations of paternal GNAS have the AHO phenotype because of haploinsufficiency of GNAS expression in bone but are not obese and are intellectually intact; their kidneys, where maternal GNAS is expressed, are normally responsive to endogenous and exogenous PTH, and hence these patients are normocalcemic and normophosphatemic. Heterozygous mutations in GNAS on either parental allele have been associated with intrauterine growth retardation, with severity greatest when the variant is in the paternal allele, suggesting that a paternally derived GNAS transcript is required for normal fetal growth. The postnatal growth retardation of PHP and PPHP is possibly the extended consequence of cartilage growth plate resistance to PTH in utero and postnatally. Patients with PHP type IA are resistant to many hormones that signal through GNAS including: TSH that may present as congenital hypothyroidism with hyperthyrotropinemia, growth hormone releasing hormone (GHRH) associated with growth hormone (GH) deficiency, luteinizing (LH) and follicle-stimulating (FSH) hormones presenting as delayed puberty, and melanocortin (MSH) associated with obesity of early onset.
Acrodysostosis is a chondrodysplasia with many features of PHP1a (short stature, obesity, brachydactyly, abnormal face with nasal and maxillary hypoplasia, hypertelorism, markedly advanced skeletal maturation) that is caused by gain-of-function variants of PRKAR1A (OMIM 188830) or PDE4D (OMIM 600129) encoding components of the signal transduction systems activated by G sα (vide supra). PRKAR1A encodes the cyclic AMP-dependent regulatory alpha subunit of PKA, the protein kinase that is downstream of G sα and cyclic AMP and whose activation leads to the cascade of intracellular signal transduction that regulates cell division, differentiation, metabolism, function, and apoptosis. Paradoxically, prolonged activation of the regulatory alpha subunit of PKA results in decline in the functional activity of the catalytic subunit of PKA. In a patient with acrodysostosis type 1 (OMIM 101800), a de novo germline gain-of-function mutation (pArg368Stop) in PRKAR1A is present that leads to functional resistance to PTH and TSH. The mutant PRKAR1A is a shortened protein whose binding avidity to the catalytic subunit of PKA is increased, because it lacks one of two cyclic AMP binding domains; hence it can only be slowly released from the catalytic subunit of PKA by cyclic AMP, thereby maintaining the catalytic subunit of PKA in the inactive state. In some patients with acrodysostosis type 1, resistance to PTH, GHRH, TSH, and the gonadotropins is present. PDE4D encodes phosphodiesterase 4D, an enzyme that hydrolyzes and inactivates cyclic AMP; hormone resistance is not present in patients with acrodysostosis type 2 (OMIM 614613) related to variants of PDE4D , although these patients are developmentally challenged. In patients with activating mutations in PDE4D , the rate of degradation of cyclic AMP is increased.
(An alternative nomenclature for PHP has been presented by the EuroPHP network and termed the inactivating PTH/PTHrP signaling disorder [iPPSD] classification. In this schema, the clinical disorders are linked to the genetic mutation in a component of the signal transduction pathways transmitting the messages of PTH and PTHrP that initiate cellular responses. For example, iPPSD1 is associated with biallelic inactivating mutations of PTHR1 leading to the usually fatal Blomstrand chondrodysplasia [OMIM 215045] of short limbs, sclerotic bones, advanced bone maturation, and facial dysmorphism associated with polyhydramnios and hydrops fetalis; Eiken syndrome [OMIM 600002] is an osteodysplasia with markedly delayed epiphyseal maturation that is also associated with biallelic mutations in PTHR1 ; Murk-Jansen chondrodysplasia [OMIM 156400] is the result of monoallelic activating mutations of PTHR1 and is characterized by small stature, short and bowed limbs, clinodactyly, abnormal face, hypercalcemia, and hypophosphatemia despite normal to low serum concentrations of PTH. Inactivating mutations of GNAS leading to PHP1a are designated iPPSD2. Methylation defects involving GNAS giving rise to PHP1b are designated iPPSD3. Heterozygous mutations in PRKAR1A [OMIM 188830] leading to deficient PKA activity result in acrodysostosis type 1 [OMIM 101800] characterized by growth retardation, variable facial dysmorphism, brachydactyly, and advanced skeletal maturation often associated with end-organ resistance to PTH and other G-protein–associated hormones are designated iPPSD4. Monoallelic variants of PDE4D [OMIM 600129] resulting in abnormalities of cyclic AMP–specific phosphodiesterase hydrolytic activity leading to acrodysostosis type 2 [OMIM 614613] whose phenotype resembles that of type 1 in association with developmental delay is termed iPPSD5 . Brachydactyly with hypertension [OMIM 112410] has been related to variants of PDE3A [OMIM 123805, chr. 12p12.2] and assigned to iPPSD6. The designation iPPSD7 is used for disorders of unknown genetic pathogenesis.)
Progressive osseous heteroplasia (POH, OMIM 166350) is one of a group of disorders associated with intra- or subdermal ossification/calcification, in addition to the AHO phenotype of PHP, that is related to inactivating variants of paternal GNAS (vide infra). POH is a disorder caused by loss-of-function mutations involving the paternal GNAS allele that is characterized clinically by dermal ossifications that begin in infancy and progress to diffuse heterotopic bone formation in skeletal muscle and deep fascia. POH is to be distinguished from other syndromes associated with ectopic calcification and ossification, including fibrodysplasia ossificans progressiva (OMIM 135100; ACVR1 —OMIM 135100) and hyperphosphatemic tumoral calcinosis (OMIM 211900; GALNT3 —OMIM 211900). Although not as yet clinically described, an inactivating mutation in LRP6 (encoding lipoprotein receptor-related protein 6—OMIM 603507) might also be associated with resistance to the biological effect of PTH. In addition to its primary role in receptor-mediated endocytosis of lipoproteins, LRP6 is essential for the movement of G sα to the plasma membrane and for its coupling to PTH1R.
Evaluation and Management
Evaluation of the neonate with hypocalcemia begins with review of the maternal preconceptual history, the conduct of gestation, and the peripartum, postnatal, and family histories followed by a comprehensive physical examination ( Fig. 20.3 ). Historical data include those related to maternal parity and complications of pregnancy, such as maternal diabetes mellitus, toxemia of pregnancy or ingestion of agents that may cause maternal hypercalcemia (excessive alkali), intrauterine growth restriction, abnormalities of delivery, neonatal sepsis, or other early postpartum illnesses. The family history is examined for members with abnormalities of mineral metabolism, such as renal calculi, rickets, or hypocalcemia (e.g., seizure disorders). The social history provides information about the socioeconomic status of the mother and her cultural milieu that may have impacted maternal diet and exposure to sunlight during gestation. Physical examination of the neonate (abnormal face, cardiac murmur consistent with congenital heart disease) may suggest a complex form of hypocalcemia. Determination of a complete blood count, serum concentrations of total calcium, Ca 2 + , magnesium, phosphate, creatinine, intact PTH, calcidiol, and calcitriol, and urinary calcium and creatinine concentrations in a spot urine should precede initial therapy of the hypocalcemic newborn whenever possible. Decreased serum concentrations of PTH are common in neonates with early-onset hypocalcemia, but persistently low PTH levels suggest impaired PTH secretion, at times because of inactivating variants of PTH or to gain-of-function mutations of CASR or GNA11 . High PTH concentrations are present in patients with vitamin D deficiency or insensitivity, PTH resistance caused by loss-of-function mutations in PTH1R , GNAS (PHP), or PRKAR1A (acrodysostosis type 1) or impaired renal function. Low levels of calcidiol signify decreased maternal (and hence fetal) vitamin D stores or rarely a defect in CYP27B1 encoding vitamin D-25 hydroxylase, whereas calcitriol concentrations are inappropriately low in subjects with severely compromised renal function, hypoparathyroidism, or those with deficiency of 25OHD-1α-hydroxylase. Elevated calcitriol values suggest vitamin D resistance perhaps caused by an abnormality in VDR, a disorder that may be associated with alopecia. Skeletal radiographs may disclose osteopenia, whereas chest x-ray may not identify a thymic shadow (but is an unreliable sign in a severely ill or stressed neonate). Serum levels of calcium, Ca 2 + , phosphate, and intact PTH should be measured in the mothers of neonates with unexplained hypocalcemia as some may have unsuspected hyperparathyroidism.
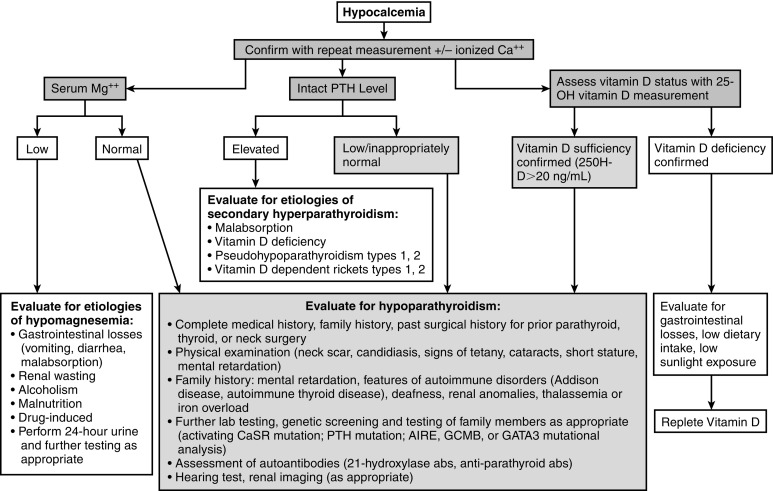
In neonates with hypocalcemia not otherwise explained, evaluation for possible DiGeorge syndrome should be undertaken—particularly when physical examination reveals an abnormal face, and a congenital anomaly of the outflow tract of the heart is present. The white blood and T (CD4) lymphocyte counts are low and the thymic shadow often absent in these subjects. The diagnosis of the DiGeorge syndrome is confirmed by the presence of a microdeletion of chromosome 22q11.2 as demonstrated by single nucleotide polymorphism based chromosomal microarray or FISH. Single nucleotide polymorphism microarray enables detection of both copy number variation (deletions or duplications) and copy-neutral structural variants, such as regions of homozygosity and uniparental disomy. Occasionally, sequence analysis of TBX1 may be needed to establish this diagnosis, if the other studies are normal. Because DiGeorge syndrome may be heritable, genetic evaluation of the parents and siblings of an affected infant is indicated. It should be noted that the majority of neonates and infants with DiGeorge syndrome are recognized primarily because of cardiac anomalies and that subjects without these lesions may not be identified until mid or late childhood or adolescence. Neonates with PHP1a may present with elevated serum levels of TSH in the neonatal metabolic screening survey but often do not have the characteristic skeletal phenotype (brachymetacarpals) of AHO; if no cause of congenital hypothyroidism is identified, the diagnosis of PHP1a should be suspected and measurement of serum calcium levels and, if appropriate, genotyping and methylation studies of GNAS are indicated.
Early neonatal hypocalcemia is often asymptomatic, but nevertheless treatment is indicated when the total serum calcium concentration is below 8 mg/dL (2 mmol/L) (Ca 2 + < 4.4 mg/dL [1.1 mmol/L]) in term infants or prematurely born neonates with birth weights over 1500 g, or in a very LBW newborn with a total calcium value less than 7 mg/dL (1.75 mmol/L) (Ca 2 + < 4.0 mg/dL [1.0 mmol/L]). Asymptomatic neonates are most easily managed by increasing the oral intake of calcium and establishing an overall ratio of calcium:phosphate intake of 4:1 (including that in feedings with a low-phosphate formula, such as Similac PM 60/40—calcium:phosphate ratio 1.6:1) with calcium glubionate or calcium carbonate administered in divided doses every 4 to 6 hours ( Table 20.3 ). Eucalcemia is almost always restored in these subjects within 3 weeks after birth and often earlier. In the hypocalcemic infant with tetany or frank seizures, 10% calcium gluconate (elemental calcium 9.3 mg/mL) at a dose of 1 to 3 mg/kg and rate of less than 1 mL/min and a total dose not to exceed 20 mg of elemental calcium/kg may be administered by intravenous infusion over 15 minutes; often seizures will cease after 1 to 3 mL of 10% calcium gluconate have been administered. Cardiac rate and rhythm must be carefully monitored to prevent bradycardia and asystole. Further intravenous bolus doses of calcium (~ 10 mg/kg at 6-hour intervals) should be used sparingly as they result in wide excursions in serum calcium values. After initial treatment of neonatal hypocalcemia, 500 mg of calcium gluconate/kg/24 hours may be administered by continuous intravenous infusion, taking care to prevent extravasation or infiltration as extracellular calcium will precipitate in soft tissues. These infants may receive supplemental oral calcium if necessary (vide supra). Depending on the cause of the hypocalcemia, supplemental vitamin D or calcitriol may also be needed. In a neonate with hypocalcemia caused by hypoparathyroidism, administration of synthetic PTH 1-34 (teriparatide) or recombinant human PTH 1-84 may occasionally be required to restore eucalcemia. Serum and urine calcium and creatinine levels should be determined frequently and treatment modified to maintain eucalcemia and the urine calcium/creatinine ratio under 0.2 in an effort to avoid iatrogenic hypercalcemia, hypercalciuria, nephrocalcinosis, and renal insufficiency. In neonates that require parenteral alimentation, 50 mg of elemental calcium/kg/24 hours should be incorporated into the infused solution; elemental phosphate must also be administered as permitted and indicated but separately from calcium.
| Content | Elemental Mineral | |
|---|---|---|
| Vitamin D | ||
| Vitamin D | ||
| Calciferol | 8000 IU/mL | |
| 50,000 IU/capsule | ||
| Calcidiol | 20 or 50 mcg/tablet | |
| Calcitriol | ||
| Rocaltrol | 1 mcg/mL (oral solution) | |
| 0.25 mcg or 0.5 mcg/capsule | ||
| Dihydrotachysterol | ||
| Hytacherol | 0.2 mg/5 mL | |
| 0.125, 0.2, 0.4 mg/tablet | ||
| Calcium | ||
| Calcium acetate | ||
| Phoslyra | 667 mg/5 mL | 667 mg/capsule |
| Calcium gluconate (iv) | 93 mg/10 mL | 93 mg/g |
| 500, 650 mg/tablet | ||
| Calcium glubionate (solution) | 64 mg/g | 115 mg/5 mL |
| Calcium carbonate | 400 mg/g | 500, 600, 750, 1250 mg/tablet |
| Titralac | 420 mg tablet | 168 mg/tablet |
| 780 mg tablet | 300 mg/tablet | |
| Os-Cal | 650 mg tablet | 260 mg/tablet (Vitamin D) |
| 1250 mg tablet | 500 mg/tablet (Vitamin D) | |
| Tums | 500 mg tablet | 200 mg/tablet |
| 750 mg tablet | 300 mg/tablet | |
| 1000 mg tablet | 400 mg/tablet | |
| 1250 mg tablet | 280 mg/tablet | |
| Caltrate | 1500 mg tablet | 600 mg/tablet (Vitamin D) |
| Calcium carbonate (suspension) | 1250 mg/5 mL | 500 mg/5 mL |
| Calcium chloride | 100 mg/mL | |
| Calcium citrate | 210 mg/g | 200 mg/tablet |
| Citracal | 950 mg tablet | 200 mg/tablet |
| Calcium lactate | 130 mg/g | 84 mg/tablet |
| Magnesium | ||
| Magnesium sulfate | 49 mg/mL (50% intramuscular solution) | |
| 0.32, 0.64, 4 mEq/mL (intravenous solution) | ||
| Magnesium oxide | 603 mg/g | 241 mg/tablet |
| Magnesium gluconate | 54 mg/g | 27 mg/tablet |
| Magnesium chloride | 120 mg/g | 64 mg/tablet |
| Phosphorus | ||
| Sodium phosphate (Phospha-Soda) | 127 mg/mL | |
| Sodium/potassium phosphate (Phos-NaK) | 250 mg/packet (powder) | |
| Potassium phosphate (Neutraphos-K) | 250 mg/packet (powder) | |
| Potassium phosphate (K-Phos Original) | 114 mg/tablet | |
| Sodium/potassium phosphate (K-Phos #2) | 250 mg/coated tablet | |
| Sodium/potassium phosphate (K-Phos Neutral) | 250 mg/tablet | |
After restoration of eucalcemia in the infant with DiGeorge syndrome, other components of this disorder must be addressed. Cardiac anomalies often require surgical correction as do palatal clefts. In affected infants who are immunocompromised and experience recurrent infections because of thymic aplasia, appropriate antiinfectious therapy is mandatory. Transplantation of fetal or cultured postnatal thymic tissue, bone marrow, or peripheral blood mononuclear cells has restored immune function in infants with this disorder. Supplemental calcitriol (20–60 ng/kg/d) and calcium are necessary for restoration and maintenance of eucalcemia in infants with hypoparathyroidism. Poor growth caused by feeding difficulties and learning disabilities caused by developmental delay must be managed on an individual basis and illustrate the need for a multidisciplinary approach to the care of these patients. Hypocalcemia caused by hypomagnesemia is managed acutely by the intravenous infusion or intramuscular injection of 50% magnesium sulfate at a dose of 0.1 to 0.2 mL/kg while monitoring cardiac status.
Hypocalcemia in the Child and Adolescent
Etiology
Causes of hypocalcemia in the child and adolescent include many that also result in neonatal hypocalcemia and are listed in Tables 20.2A, 20.2B and Fig. 20.1 . Hypocalcemia is defined by the norms of the analytical laboratory; total serum calcium and phosphate concentrations vary by age. (A general guideline for age-related total calcium concentrations is: 1–5 years: 9.4–10.8; 6–12 years: 9.4–10.2; > 20 years: 8.8–10.2 mg/dL.) Total calcium levels are low in the hypoalbuminemic patient—a correction for hypoalbuminemia may be calculated by adding 0.8 mg/dL to the recorded total calcium concentration for every decrease in albumin concentration of 1 g/dL. Thus it is appropriate to measure both total and Ca 2 + values when evaluating the hypocalcemic child. It is particularly important to monitor Ca 2 + values in the hypocalcemic critically ill child. In the very ill child/adolescent, a low total calcium value may at times be related to hypoproteinemia or to altered acid-base balance and variable binding of calcium to serum proteins, but it may also be caused by impaired secretion of PTH related to hypomagnesemia or to decreased synthesis of vitamin D and consequent subnormal intestinal absorption of calcium. Mechanistically, hypocalcemia develops as a consequence of either too little inflow of calcium from the gastrointestinal tract, bone, or kidney into the extracellular and vascular spaces or excessive loss of calcium from these spaces into urine, stool, or bone. Thus hypocalcemia may be caused by decreased intake or absorption or excessive loss of calcium, decreased production of bioactive PTH because of congenital abnormalities of parathyroid gland development or PTH synthesis or of the CaSR, destruction of parathyroid glands by autoantibodies, metal overload (copper, iron), surgical or radiation insults, granulomatous infiltration, or to impaired cellular responsiveness to PTH. Restricted exposure to sunlight or reduced intake, absorption, metabolism, or activity of vitamin D leads to hypocalcemia. Hypomagnesemia impairs the secretion (but not the synthesis of PTH) and blunts tissue responsiveness to PTH. Hypocalcemia occurs after exposure to a number of drugs and medications. Thus hypocalcemic tetany may develop after the administration of phosphate containing enemas by rectum or laxatives by mouth. At times, the hypocalcemic child or adolescent may be asymptomatic and identified by chemical screening for an unrelated problem or may present with intermittent muscular cramping either at rest or during exercise (when the increase in systemic pH caused by hyperventilation lowers still further the concentration of Ca 2 + ); paresthesias of fingers, toes, or circumoral regions; tetany (carpopedal spasm, laryngospasm, bronchospasm); or seizures (grand mal, focal, petit mal, adynamic or syncopal). Prolonged and severe hypocalcemia may lead to congestive heart failure. Physical examination often reveals a positive Chvostek (twitching of the cheek upon tapping of the ipsilateral seventh cranial nerve) and/or Trousseau sign (carpopedal spasm) and hyperreflexia. However, a positive Chvostek sign is commonly present in normal adolescents.
Hypoparathyroidism may occur as a solitary disorder, as part of a multidimensional autoimmune polyendocrinopathy, or as one manifestation of a group of complex congenital anomalies (as described in the section on neonatal hypocalcemia). Hypoparathyroidism may be the consequence of total thyroidectomy performed to excise a malignant thyroid lesion because of inadvertent removal of the four parathyroid glands or insults to their arterial blood supply. There are sporadic and familial forms of hypoparathyroidism; when familial, hypoparathyroidism may be transmitted as an autosomal dominant, autosomal recessive, or X-linked recessive trait (see Tables 20.2A, 20.2B and see Fig. 20.1 ). Abnormalities in the development of the parathyroid glands, transcription of PTH , and in processing of the translated product have been associated with inherited forms of hypoparathyroidism. Familial isolated/autosomal dominant dyshormonogenic hypoparathyroidism may be caused by a monoallelic T ⇒ C transition in codon 18 (p.Cys18Arg) of the 25 aa signal peptide of preproPTH that impairs efficient transport of protein from the ribosome and interaction of preproPTH with the signal recognition particle, movement of the precursor peptide into and exit from the rough endoplasmic reticulum, its cleavage by a signal peptidase, and its incorporation into a secretory granule. Familial isolated/autosomal recessive dyshormonogenic hypoparathyroidism has also been associated with a homozygous G ⇒ C transversion in nucleotide 1 of intron 2 of PTH within the signal sequence that prevented normal cleavage of preproPTH and decreased secretion of PTH. Occasionally, isolated hypoparathyroidism may be found in patients with deletion of chromosome 22q11.2 without other signs or symptoms of the DiGeorge or related syndromes.
Autosomal dominant hypocalcemia type 1 (OMIM 601198) is caused by heterozygous gain-of-function mutations in the extracellular, transmembrane, and intracellular domains of CASR that transcribe a CaSR that is not intrinsically constitutively active but is exceptionally sensitive to and easily activated by very low serum Ca 2 + concentrations may be identified in infancy or in older subjects ( Fig. 20.4 ). In this disorder, even at hypocalcemic levels, Ca 2 + binds avidly to the CaSR and activates phospholipase C-β1 increasing cytosolic levels of inositol phosphate and Ca 2 + and stimulating the mitogen-activated protein kinase (MAPK) signal transduction pathway in parathyroid chief cells, thus suppressing PTH synthesis and secretion, and in the kidney, there decreasing renal tubular reabsorption of calcium and magnesium, leading to urinary wasting of these cations (hypercalciuric hypocalcemia) and decreased urinary concentrating ability. In these subjects, serum levels of phosphate are increased and magnesium values decreased; PTH concentrations are low or inappropriately normal. Autosomal dominant hypocalcemia type 2 is caused by heterozygous variants of GNA11 (OMIM 139313) that encodes a G-protein alpha subunit (Gα11) that initiates intracellular signal transduction after binding of Ca 2 + to the CaSR. Its clinical and biochemical manifestations are similar to those caused by variants of CASR with seizures, laryngospasm, hypocalcemia, hyperphosphatemia, and relatively low serum levels of PTH. Because patients with autosomal dominant hypocalcemia frequently have symptomatic hypocalcemia, such as tetany and seizures, and are very sensitive to vitamin D, the dose of calcitriol must be limited to that which raises serum calcium values to asymptomatic values even if not within the normal range as larger doses lead to hypercalciuria (sometimes even when serum calcium levels remain subnormal), nephrocalcinosis, and functional renal insufficiency. Administration of recombinant human (rh)PTH 1-34 (teriparatide) restores calcium homeostasis in this disorder but does not necessarily prevent nephrocalcinosis. However, recombinant human PTH 1-34 is not routinely administered to children because experimentally, there is an increased incidence of osteosarcoma in young rats receiving very large amounts of this agent, although primates appear to be less susceptible to PTH-induced bone tumor formation than do rodents. Development of stimulatory autoantibodies to the CaSR results in an acquired variant of spontaneous hypoparathyroidism that may be isolated or part of a complex autoimmune endocrinopathy. Indeed, in perhaps as many as one-third of patients with acquired isolated idiopathic hypoparathyroidism, antibodies directed against epitopes in the extracellular domain of the CaSR may be present. This form of acquired hypoparathyroidism may be reversible, as these antibodies do not destroy the parathyroid glands. In patients with other forms of autoimmune hypoparathyroidism, the antibodies are cytotoxic and accompanied by lymphocytic infiltration, atrophy, and fatty replacement of parathyroid tissue.
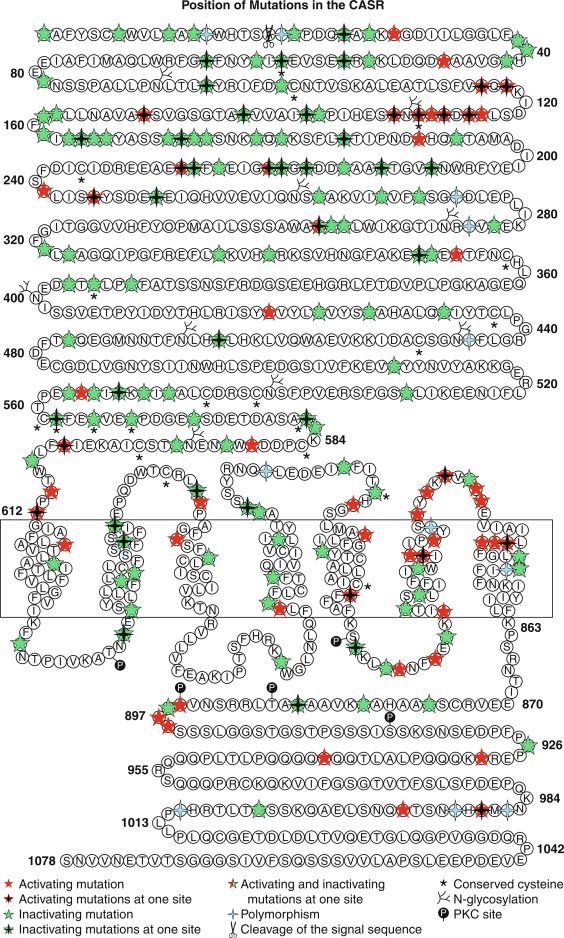
In mid-childhood and adolescence, acquired hypoparathyroidism may be a delayed or late manifestation of a genetic variant (e.g., the DiGeorge syndrome), but it may also be the result of destruction of the parathyroid glands by autoimmune disease or surgical removal or operative trauma to the vascular supply of these structures. Unusual causes of acquired hypoparathyroidism in this age group include infiltration by iron (hemochromatosis, excessive blood transfusions) or copper (Wilson hepatolenticular degeneration), granulomatous diseases, or radiation (mantle radiation for Hodgkin/non-Hodgkin lymphoma or radioiodine therapy of hyperthyroidism).
Autoimmune hypoparathyroidism may occur as an isolated disorder or as part of the complex of autoimmune polyendocrinopathy syndrome type I (APS1). Development of an autoimmune endocrinopathy is initiated by failure of recognition of a peptide specific for a target organ by a subgroup of regulatory T cells that ordinarily recognize that peptide. When immunological tolerance for that peptide is lost, clones of CD4 regulatory T cells for the peptide expand; type 1 helper T cells secrete inflammatory cytokines, such as interferon-γ (IFN-γ), whereas type 2 helper T cells stimulate B cell function and lead to autoantibody-mediated inflammation. Loss of immune tolerance may be the consequence of the postinfectious inflammatory state caused by activation of the innate immune system or caused by a gene mutation that depresses immune tolerance and permits expansion of a CD4 + regulatory T cell clone after exposure to quantitatively small amounts of antigen. Genetic variations within the major histocompatibility complex (human leukocyte antigen [HLA]-DQ, HLA-DR) that determine peptide (antigen) presentation to CD4 + regulatory T cells join with genetic abnormalities in immune regulation to induce autoimmune disease. In 30% of patients with isolated idiopathic hypoparathyroidism the disorder is caused by antibodies to the extracellular domain of the CaSR and thus functionally inhibitory but potentially reversible; in approximately 33% of patients serum antibodies to other components of the parathyroid chief cell may be present. There are several types of autoimmune polyendocrinopathy syndromes but only in type I is hypoparathyroidism a major component.
APS1 (OMIM 240300) is an autosomal recessive disorder with the classic triad of autoimmune polyendocrinopathy, mucocutaneous candidiasis, and ectodermal dystrophy (APECED). It is characterized by childhood-onset of multiple autoimmune systemic disorders and endocrinopathies including hypoparathyroidism, primary adrenal insufficiency, and chronic mucocutaneous candidiasis, and less often pernicious anemia, hepatitis, primary ovarian and testicular failure, keratitis, retinitis, alopecia, and reversible metaphyseal dysplasia. Autoimmune polyendocrine syndrome1 is caused by loss-of-function variants of autoimmune regulator ( AIRE , OMIM 607358). In a Finnish cohort of 91 patients with APS1/APECED, the cardinal manifestations were mucocutaneous candidiasis involving the nails and mouth occurring in 100% of patients—often in the first 2 years of life, hypoparathyroidism, and hypoadrenocorticism both of which illnesses developed in 80% to 90% of affected subjects. Hypoparathyroidism occurred most often between 2 and 10 years and hypoadrenocorticism developed between 5 and 15 years of age. Almost all females with APECED developed hypoparathyroidism whereas 80% of affected males did so. Hypomagnesemia, often severe and recalcitrant to therapy, was common in patients with hypoparathyroidism caused by APECED. The most frequent presenting manifestations of APECED were mucocutaneous candidiasis—60%, hypoparathyroidism—32%, and hypoadrenocorticism—5%; the disease first became apparent between 2 months and 18 years of age. However, 10% of patients presented with another manifestation of APECED. In addition to hypoparathyroidism and hypoadrenocorticism, other endocrinopathies encountered in APECED included autoimmune oophoritis leading to ovarian failure (70%), orchitis resulting in testicular failure (30%), diabetes mellitus (30%), thyroiditis (30%), and hypophysitis (4%). Besides mucocutaneous candidiasis, dermatological manifestations and complications of APECED in the Finnish cohort included alopecia (40%), vitiligo (30%), and rashes with fever (15%). Keratoconjunctivitis developed in 20% of affected subjects, pernicious anemia in 30%, hepatitis in 20%, and chronic diarrhea in 20%. In a Norwegian population of 36 patients with APECED, 13 had clinical evidence of disease at or before 5 years of age and an additional 15 subjects presented at or before 15 years of age. In a Russian population of 46 patients, mucocutaneous candidiasis was present in 70%, hypoparathyroidism in 83%, and adrenal insufficiency in 54%. Other common problems were alopecia (27%), thyroid dysfunction (20%), and malabsorption syndromes (18%). Unusual findings in this cohort were retinitis pigmentosa and metaphyseal dysplasia—both occurring in 7% of patients. Later manifestations of APECED include esophageal and oral squamous cell carcinoma, asplenia, and interstitial nephritis. The diagnosis of APECED usually requires the presence of two of three of its major diseases (mucocutaneous candidiasis, hypoparathyroidism, hypoadrenocorticism), but occasionally chronic candidiasis, hypoparathyroidism or primary adrenal insufficiency alone may be the only manifestation of an inactivating mutation in AIRE.
APS1 is the result of homozygous or compound heterozygous loss-of-function mutations in AIRE , a 14 exon gene encoding a 545 aa protein with two zinc-finger motifs that is expressed in nuclei of thymic medullary epithelial cells and in lymph nodes, spleen, and monocytes. AIRE also serves as an E3 ubiquitin ligase, an essential component of the ubiquitin-proteasomal system for protein modification and destruction involved in cellular division and differentiation, protein transport, and intracellular signaling. Structurally, AIRE contains two plant homeodomains—a sequence of amino acids composed of an octet of cysteines and histidines that coordinate two zinc ions; the first plant homeodomain is essential for the E3 ubiquitin ligase activity of the protein and the second plant homeodomain is required for its transcription-regulating action. AIRE also contains a SAND domain, a structure that permits it to bind DNA. AIRE enables thymic cells (T lymphocytes) to distinguish between normal self-antigens and proteins that are foreign to the host. AIRE does so by inducing the expression of normal self-antigens in the thymus and then eliminating the clonal subsets of T lymphocytes that recognize them thereby establishing self-tolerance. To develop an immune system that is capable of distinguishing between “self” and “foreign” antigens, thymic stromal medullary epithelial cells locally transcribe the majority of protein encoding genes expressed in various peripheral tissues to permit their recognition by developing T cells before the release of these T cells from the thymus and thus prevent an autoimmune response when these proteins are encountered by T cells in the periphery. The autoimmune regulatory gene AIRE enables the thymic transcription and expression of these genes by interacting with nuclear transporters (e.g., exportin), chromatin binding proteins (e.g., histones), transcription factors and postinitiation transcription processes (e.g., CREB; complex of DNA-dependent protein kinase-polymerase–topoisomerase), and pre-messenger (m)RNA splicing and processing factors. AIRE does not function as a specific transcription factor. Inasmuch as nuclear AIRE binds to the unmethylated tails of histone-3 in chromatin, it has been proposed that AIRE activates quiescent genes by attracting to their site the transcription apparatus that enables initiation of transcription, elongation, and pre-mRNA processing. In the Finnish and Russian populations with APS1, the most common loss-of-function mutation in AIRE was a homozygous truncating mutation at codon 257 (p.Arg257Ter). Pathogenic mutations in AIRE detected in patients with APECED include missense, nonsense (p.Arg139Ter), insertions, and deletions (e.g., 13 base pair deletion—964del13, NT 1094, exon 8) that alter the subcellular distribution of AIRE and/or decrease its transcriptional activation capacity and/or its E3 ubiquitin ligase activity. AIRE also stimulates development of thymic FoxP3 + regulatory T cells (Treg) that are able to inhibit autoreactive T cells.
Thymic levels of AIRE are lower in normal postpubertal estrogenized females than those in normal postpubertal masculinized males, possibly accounting in part for the increased susceptibility of females to the later development of autoimmune disorders. Despite thymic expression of three copies of AIRE , subjects with trisomy 21 (Down syndrome) have decreased thymic tissue levels of AIRE, thereby perhaps predisposing this patient population to the development of autoimmune diseases. When AIRE is inactivated by biallelic loss-of-function mutations (e.g., p.Arg257X, p.Cys322del13), self-recognition of T cells is lost; normal self-proteins are not distinguishable from foreign proteins, and thus a destructive (auto)immune response to self-antigens is initiated. (Some variants of AIRE are transmitted as autosomal dominant or dominant negative disorders. ) Patients with mutations of AIRE and autoimmune polyendocrinopathy type 1 develop organ-related antibodies to cytoplasmic proteins (e.g., to parathyroid-specific NALRP5 [NACHT-domain, leucine-rich repeat, pyrin domain-containing protein 5, OMIM 609658] that are often demonstrable in patients with hypoparathyroidism). The presence of autoimmune polyendocrinopathy syndrome type 1 is established by the clinical findings, associated endocrinopathies, and measurable autoantibodies to other endocrine tissues (e.g., glutamic acid decarboxylase 65 [type 1 diabetes mellitus], 21-hydroxylase [adrenocortical failure]), and nonendocrine tissues (e.g., retina) and confirmed by genotyping of AIRE . Patients with endocrinopathies attributed to autoimmune polyendocrinopathy type 1 are treated appropriately for the hormone deficiency(ies) present, as well as for the other manifestations of this illness. Patients with hypoparathyroidism require supplemental vitamin D (calcitriol), calcium, and magnesium; recombinant PTH 1-34 and PTH 1-84 have been used in adults for treatment of hypoparathyroidism (although not US Food and Drug Administration [FDA] approved for use in children because of concern about their oncogenic potential). All asymptomatic first-degree relatives of the patient with autoimmune polyendocrinopathy type 1 should be surveyed for the presence of a presymptomatic stage of this illness.
Autoimmune polyendocrine syndrome type II is a polygenic autoimmune disorder manifested by adrenalitis, thyroiditis, and type 1 diabetes mellitus (and in some patients pernicious anemia, hypergonadotropic hypogonadism, hepatitis, celiac disease, myasthenia gravis, alopecia, and vitiligo) and is related to variants in major histocompatibility complexes, for example, DR3-DQ2, DR4-DQ8, DRB1*04, DQB1*02, and others. Hypoparathyroidism does not occur in patients with this disorder. The syndrome of Immunodeficiency, Polyendocrinopathy, and Enteropathy–X-linked (IPEX – OMIM 34790) is associated with the early (neonatal and early childhood) onset of autoimmune enteropathy, insulin-dependent diabetes mellitus, autoimmune thyroid disease, eczema, glomerulonephritis, thrombocytopenia, and hemolytic anemia that occurs only in males because it is the result of loss-of-function mutations in FOXP3 (OMIM 300292). FOXP3 is a transcription factor that is essential for normal development of naturally occurring T regulatory cells that maintain self-tolerance. PHP is a disorder with variable clinical manifestations associated with resistance to the action of PTH whose clinical phenotypes and genetic variabilities have been discussed earlier.
Deficiency of vitamin D intake, aberrant metabolism of cholecalciferol, or decreased biological responsiveness of the VDR may result in hypocalcemia. In the subject with skeletal demineralization caused by marked vitamin D deficiency, serum calcium concentrations may fall precipitously after administration of even small amounts of vitamin D as renewed mineralization of bone matrix caused by accelerated osteoblastic activity consumes calcium and phosphate (the “hungry bone” syndrome). After parathyroidectomy for primary hyperparathyroidism, calcium concentrations often decline rapidly by the same mechanism. Drugs that inhibit PTH secretion (excessive magnesium), osteoclast resorption of bone (bisphosphonates), or renal resorption of calcium (furosemide) may lead to hypocalcemia. Intravenous infusion or rectal administration of phosphate (in enemas), acute cellular destruction by tumor cell lysis or rhabdomyolysis, and acute and chronic renal failure increase serum phosphate levels and lead to reciprocal decline in calcium values. Serum calcium concentrations decline in patients receiving multiple transfusions of citrated blood or during plasmapheresis. In subjects with acute pancreatitis, calcium complexed with free fatty acids generated by pancreatic lipase is deposited in necrotic tissue. Acute severe illness of diverse pathogenesis is often associated with hypocalcemia; this has been attributed to hypoalbuminemia, hyperphosphatemia, alkalosis, functional hypoparathyroidism, hypercalcitonemia, hypomagnesemia, vitamin D deficiency, decreased calcitriol synthesis, alkalosis and increased serum concentrations of free fatty acids (the latter increase binding of Ca 2 + to albumin), increased cytokine activity, and administered medications (e.g., aminoglycosides, citrated blood, calcium chelators, fluoride).
Evaluation
Fig. 20.3 outlines the evaluation of the child/adolescent with hypocalcemia. Hypocalcemia may be asymptomatic until detected by a multiassay chemical profile obtained for another purpose. A prolonged QT interval noted by electrocardiography obtained for evaluation of a functional heart murmur or arrhythmia suggests the presence of hypocalcemia. Roentgenograms obtained for evaluation of abdominal pain or following a head injury may reveal nephrocalcinosis, renal calculi, or calcification of the basal ganglia of the brain, respectively; such findings merit measurement of the serum calcium concentration. The major symptoms and physical signs of hypocalcemia depend on its rapidity of onset and depth. The more quickly is the onset of hypocalcemia and the lower the calcium value achieved, the more substantial are the symptoms. Symptoms may include paresthesias (numbness and tingling of the hands, feet, perioral region), muscular hyperexcitability, including cramping, carpopedal spasm (flexion of the elbow and wrist, adduction of the thumb, flexion of the metacarpal/metatarsal-phalangeal joints, and extension of the interphalangeal joints), tetany—particularly during vigorous exercise—occasionally generalized seizures, bronchospasm, and at times neuropsychiatric symptoms. (Laryngospasm is a life-threatening complication of hypocalcemia.) Review of the past medical and family histories may reveal symptoms consistent with or illnesses associated with hypocalcemia (recurrent infections, congenital cardiac anomalies, surgical procedures in the neck, cervical radiation, infiltrative diseases), or may identify related members with hypoparathyroidism, dysmorphic physical characteristics, autoimmune endocrinopathies, or hypomagnesemia.
Physical examination may disclose characteristic abnormalities in children with PHP type IA in whom the phenotype of AHO is present (short stature, round face, subcutaneous hard nodules, brachymetacarpals), the DiGeorge syndrome (atypical face, recurrent infections, cardiac murmur), or familial APS1 (chronic mucocutaneous candidiasis and other ectodermal abnormalities, such as vitiligo, alopecia, keratoconjunctivitis), whereas rachitic deformities in the hypocalcemic child imply the presence of a form of hypovitaminosis D. Most commonly, however, the physical examination reveals no striking abnormality in the hypocalcemic child other than those of increased neuromuscular irritability, such as hyperreflexia, positive Chvostek sign (twitching of the circumoral muscles when tapping lightly over the seventh cranial nerve) or Trousseau sign (carpal pedal spasm when maintaining the blood pressure cuff 20 mm Hg above the systolic blood pressure for 3 minutes) and occasionally cataracts, papilledema, or abnormal dentition. (Tetany also occurs in subjects with hypo- and hypernatremia, hypo- and hyperkalemia, and hypomagnesemia, and a positive Chvostek sign may be found in many normal adolescents.)
Laboratory evaluation of hypocalcemia includes measurements of serum concentrations of total calcium, Ca 2 + , phosphate, magnesium, PTH, and 25-hydroxyvitamin D, and determination of the urine calcium excretion. In most hypocalcemic patients, urine calcium excretion is low; however, if the urine calcium excretion is inappropriately normal or high, disorders, such as autosomal dominant hypocalcemia type 1 (gain-of-function mutation in CASR or GNA11 ) may be considered; these genes may then be analyzed or antibodies to CaSR determined as clinically indicated. Antibodies to the CaSR have been detected in approximately 50% to 80% of patients with autoimmune polyendocrinopathy type 1. The child/adolescent with hypocalcemia, hypocalciuria, hyperphosphatemia, and low or undetectable serum PTH concentration (and normal or only slightly low serum magnesium level) likely has hypoparathyroidism because of a primary defect in PTH synthesis or secretion related to congenital malformation or acquired destruction of the parathyroid glands. Patients with hypoparathyroidism often have low serum concentrations of calcitriol, normal levels of calcidiol, decreased excretion of urinary nephrogenous cyclic AMP, and increased renal tubular reabsorption of phosphate. The DiGeorge syndrome may be identified by microarray or FISH analysis of chromosome 22q11.2 using probes directed to the deleted segment; occasionally TBX1 genotyping may be helpful. Analysis of CASR , GNA11 , PTH , GCM2 , FHL1 , and other genes associated with isolated or syndromic hypoparathyroidism ( TCBE , GATA3 ) may be indicated in the appropriate clinical context. The diagnosis of APS1 is based on clinical and laboratory findings and genotyping of AIRE . The presence of two of three of its major manifestations (candidiasis and/or ectodermal dystrophy, hypoparathyroidism, hypoadrenocorticism) are accepted criteria for its clinical diagnosis, but isolated hypoparathyroidism, adrenal insufficiency, or chronic mucocandidiasis may occasionally be its only manifestation. Antibodies to the CaSR or other cellular components of the parathyroid glands, the adrenal glands (side-chain cleavage, 21-hydroxylase, 17α-hydroxylase enzymes), neurotransmitters (aromatic L-amino acid decarboxylase, tryptophan hydroxylase), and IFNs-α and -ω may be determined; antibodies to IFN-ω are commonly present in patients with APS1/APECED. Genotyping of AIRE and identification of the mutation(s) confirm the diagnosis of APECED. There is wide variability in the clinical expression of APECED both between families and among siblings because the phenotype is not directly related to the genotype. In patients with isolated, idiopathic hypoparathyroidism, a search for antibodies to the CaSR and/or to parathyroid tissue is warranted. In hypomagnesemic subjects, magnesium and PTH levels are quite low, and PTH secretion increases rapidly after intravenous administration of magnesium. Primary hypomagnesemia should be considered when hypocalcemia and hypercalciuria coincide and serum PTH and magnesium values are low; urinary magnesium excretion should then be quantitated and CLDN1 6 (OMIM 603959) genotyped. Because hypomagnesemia may also be caused by a selective small intestinal defect in magnesium absorption, mutations in the transient receptor potential, cation channel, subfamily M, member 6 ( TRPM6 , OMIM 607009) should be examined as warranted. Primary hypomagnesemia must be differentiated from that caused by the Gittleman and Bartter syndromes (vide infra).
An elevated serum concentration of PTH in a hypocalcemic subject suggests that the patient is either secreting an abnormal PTH molecule or is resistant to PTH or there is a compensatory (secondary) PTH secretory response to hypocalcemia (see Fig. 20.3 ). Physicochemical characterization of the PTH molecule and analysis of PTH (OMIM 168450) enable one to define the abnormality in PTH synthesis, posttranslational processing, secretion, or activity leading to the functionally hypoparathyroid state. Analysis of GNAS (OMIM 139320) and its pattern of imprinting permits identification of the specific genetic defect in the majority of patients with clinical PHP type IA and PPHP. Skeletal and renal responsiveness to PTH may be assessed if warranted by measurement of changes in serum calcium, Ca 2 + , phosphate, cyclic AMP, and calcitriol concentrations and urinary nephrogenous cyclic AMP and phosphate excretion following administration of biosynthetic PTH 1-34 (Elsworth-Howard test). In the normal subject and in the patient with primary hypoparathyroidism, urinary cyclic AMP excretion increases 10- to 20-fold and that of phosphate severalfold; in the patient with PHP types IA and IB there is less than a threefold increase in the excretion of cyclic AMP after administration of PTH 1-34 . The diagnosis of PHP type IB may be established by examining the imprinting patterns of GNAS (i.e., the methylation status of the differentially methylated regions of GNAS ) and analysis of STX16 (OMIM 603666) and the 5′ sequences of GNAS as indicated. Acrodystostosis 1 and 2 (OMIM 101800;614613) are skeletal dysplasias with hormone resistance that should also be considered when evaluating a hypocalcemic infant with inappropriately elevated serum levels of PTH. This chondrodystrophy is attributable to inactivating variants of PRKAR1A (OMIM 188830) or PDE4D (OMIM 600129). PRKAR1A encodes a component of PKA—the major mediator of postreceptor intracellular signal transduction; PDE4D encodes a cyclic AMP-specific phosphodiesterase, loss of which alters cyclic AMP function. In the child with vitamin D deficiency, serum levels of calcidiol are low. In patients with decreased renal 25OHD 3 -1α-hydroxylase activity, serum concentrations of calcidiol are normal whereas those of calcitriol are inappropriately low. Increased concentrations of calcitriol suggest the presence of a defect in the nuclear VDR. The patient with renal failure is recognized by an increased serum creatinine value. Other findings in hypocalcemic subjects include prolongation of the QT interval by electrocardiography and calcification of the basal ganglia by cranial computerized tomography.
Management
The primary goal in the care of the hypocalcemic child and adolescent is to increase serum calcium concentrations to levels at which the patient is asymptomatic and as close to the lower range of normal as possible without inducing hypercalcemia; the secondary goal is to identify the cause of hypocalcemia as quickly as possible to provide disease-specific management. The overall aims of management of hypocalcemia in general and hypoparathyroidism specifically are to: (1) ameliorate the signs and symptoms of hypocalcemia, (2) to prevent hypercalcemia by (3) maintaining the serum concentration of calcium at or slightly below the lower range of normal in order (4) to maintain the calcium × phosphate product below 55 mg/dL and thus (5) to prevent hypercalciuria and (6) calcium/phosphate precipitation and extraskeletal calcifications.
Asymptomatic hypocalcemia (total calcium > 7.5 mg/dL = 1.88 mmol/L) may not require immediate intervention. With lower serum calcium levels or when hypocalcemia is symptomatic (tetany, seizures, laryngospasm, bronchospasm), acute management may require the intravenous administration of 10% calcium gluconate (93 mg of elemental calcium/10 mL vial) at a slow rate (not > 2 mL [1.86 mg of elemental calcium]/kg over 10 minutes) while closely monitoring pulse rate (and the electrocardiographic QT interval). Acutely, intravenous administration of calcium is intended to ameliorate the more serious consequences of hypocalcemia, such as seizures, bronchospasm, or laryngospasm, not to restore and maintain the eucalcemic state. Intravenously, calcium should not be administered with phosphate or bicarbonate, because these salts may coprecipitate. Extravascular extravasation of calcium is to be avoided because it may precipitate and cause local tissue injury. After the acute symptoms have resolved, calcium gluconate (10 mL = 93 mg of elemental calcium in 100 mL 5% dextrose/0.25 normal saline) may be infused intravenously at a rate sufficient to maintain calcium levels in the asymptomatic low-normal range while the cause of the hypocalcemia is identified and more specific therapy for persistent hypocalcemia prescribed. In the child with marked hyperphosphatemia as a cause of hypocalcemia, in addition to parenteral calcium administration infusion of normal saline sufficient to maintain urine output at or above 2 mL/kg per hour is necessary. Frequent measurement of serum calcium and phosphate concentrations permits rapid adjustment of fluid and electrolyte therapy. The evaluation should proceed as rapidly as possible and oral therapy begun reasonably quickly.
After stabilization, patients with hypoparathyroidism or PHP may be treated with calcitriol (20–60 ng/kg/d in 2 equally divided doses) and supplemental calcium (calcium glubionate, calcium citrate, calcium carbonate), 30 to 75 mg elemental calcium/kg/d in 4 divided doses daily often with meals—(see Table 20.3 ) to restore and maintain eucalcemia. The serum calcium concentration should be maintained within the low-normal range. Each patient must be carefully monitored to avoid hypercalcemia, hypercalciuria (calcium excretion greater than 4 mg/kg/24 h), nephrocalcinosis, and nephrolithiasis. Basal and periodic measurements of serum concentrations of calcium, phosphate, magnesium, and creatinine and urinary calcium and creatinine excretion (every 3 months) are advisable. Twice yearly, renal sonograms are also suggested to identify the development of nephrocalcinosis in its earliest stage. Children with autosomal dominant hypocalcemia caused by gain-of-function mutations in CASR (OMIM 601199) or GNA11 (OMIM 139320) are extremely sensitive to vitamin D and its metabolites. Even small doses of calcitriol may lead to hypercalciuria with minimal increase in serum calcium levels; in that instance, addition of hydrochlorothiazide (0.5–2.0 mg/kg/d) may increase renal tubular reabsorption of calcium and lower the calcitriol requirement. Administration of recombinant human PTH 1-34 has been beneficial in individual subjects with hypoparathyroidism. In adults and children with hypoparathyroidism caused by a variety of causes, the use of rhPTH 1-34 (0.4–0.5 mcg/kg every 12 hours subcutaneously) together with calcium carbonate and vitamin D (1200 mg/day of elemental calcium and 800 IU/day of cholecalciferol in 4 equally divided doses) has proven effective and safe for as long as 3 years. rhPTH 1-34 led to acceleration of bone turnover as reflected by increases in serum alkaline phosphatase and osteocalcin and urinary excretion of pyridinoline and deoxypyridinoline, while accrual of bone mineral mass, linear growth, and weight gain were not adversely impacted in treated children. In six children with primary hypoparathyroidism, subcutaneous administration of rhPTH 1-34 (12.5 mcg twice daily) was well tolerated and allowed withdrawal or lowering of the doses of supplemental calcium and calcitriol. Continuous infusion of rhPTH 1-34 has also proven effective in maintaining reasonable serum calcium concentrations in adults with postsurgical hypoparathyroidism with fewer fluctuations in calcium levels and lower urine calcium values when compared with twice daily subcutaneous injections of rhPTH 1-34 . However, at present rhPTH 1-34 is not indicated for routine treatment of children with hypoparathyroidism because of concerns about its link to osteosarcoma. In adults, rhPTH has also been used in the treatment of hypoparathyroidism with substantial decline in supplemental calcium and calcitriol doses and stabilization of serum and urine calcium values over 6 years. In addition, bone turnover markers reflected increase in skeletal formation documented by measurement of bone mineral density (BMD) of the lumbar spine. The relationship of rhPTH 1-34 to bone tumor formation, if any, is unknown at present.
When undertaking total thyroidectomy for treatment of a thyroid malignancy, consider the possible effects of this procedure upon function of the parathyroid glands. Before surgery, serum concentrations of calcium, magnesium, phosphate, and PTH should be recorded. During the operation, efforts to identify and preserve the parathyroid glands and their blood supply should be undertaken. Physical examination seeking signs of hypocalcemia (Chvostek) and serial measurements of calcium, magnesium, and PTH should begin immediately after completion of surgery and continue as clinically indicated.
Because PHP type IA is associated with resistance to a number of peptide hormones that act through GPCR, periodic assessment of pituitary-thyroid and pituitary-ovarian function and GH secretion is necessary and hormone replacement therapy begun as indicated. In general, the short stature of PHP type IA reflects the AHO phenotype and not GH deficiency. However, PHP-associated hyposomatotropism may be responsive to exogenous recombinant human GH. Transient hypoparathyroidism of infancy may be the initial manifestation of later onset hypoparathyroidism; thus assess calcium homeostasis in such subjects throughout childhood. Patients with apparently isolated hypoparathyroidism of unknown etiology should be reevaluated periodically to identify the development of autoimmune disorders in the patient or family. Assessment of thymic function is important in those subjects with findings suggestive of the DiGeorge syndrome. The management of patients with vitamin D deficiency or resistance is discussed in subsequent sections.
When hypomagnesemia is symptomatic, administration of magnesium sulfate parenterally may be necessary (50% solution, 0.1–0.2 mL/kg intramuscularly, repeated after 12–24 hours if needed). The patient with primary hypomagnesemia may require daily parenteral (intramuscular, intravenous) doses of magnesium sulfate to prevent tetany, seizures, and other neurological symptoms (slurred speech, choreoathetoid movements, weakness), and to enable normal growth and development. Calcitriol alone raises serum calcium levels in hypomagnesemic subjects but is ineffective in the prevention of tetany. Continuous overnight nasogastric infusion of magnesium may help alleviate the gastrointestinal side effects of multiple large doses of oral magnesium. More mild and transient forms of hypomagnesemia may be treated with oral magnesium gluconate or tribasic magnesium citrate (see Table 20.3 ).
Hypercalcemia
Hypercalcemia is present when the serum concentration of ionized calcium is above the normal range for age. The total serum calcium concentration reflects both the serum protein level and serum pH; generally, both total and ionized calcium concentrations should be routinely measured when hypercalcemia is suspected. Hypercalcemia may be considered mild when the total serum calcium concentration is above the upper limit of the normal range and below 12 mg/dL (3.0 mmol/L), moderate with total calcium values of 12 to 14 mg/dL (3.00–3.50 mmol/L), or severe when the total serum calcium value exceeds 14 mg/dL (3.50 mmol/L). Pathophysiologically, hypercalcemia may be the result of excessive intestinal absorption of ingested calcium (e.g., vitamin D excess), increased reabsorption of skeletal calcium caused by enhanced PTH action or a lytic bone lesion, or increased renal tubular reabsorption of calcium caused by a medication, such as a thiazide diuretic. Thus hypercalcemia may be caused by aberrations that are PTH-dependent or -independent.
Hypercalcemia in the Neonate and Infant
Hypercalcemia in neonates and very young infants is defined as total blood calcium concentration above 10.8 to 11.3 mg/dL (2.70–2.83 mmol/L) and Ca 2 + greater than 5.92 to 6.40 mg/dL (1.48–1.60 mmol/L) (depending on the analytical laboratory). However, substantial symptoms and signs of hypercalcemia (e.g., lethargy, anorexia, emesis, constipation, polyuria, dehydration, gastroesophageal reflux and emesis, constipation, and lethargy and hypotonia or irritability and seizures) may not occur until the total serum calcium level exceeds 12.5 to 13 mg/dL (3.12–4.25 mmol/L). Hypercalcemic infants frequently are polyuric because of renal resistance to antidiuretic hormone and may become dehydrated if fluid intake is restricted. Because of the vasoconstrictive effect of calcium, the hypercalcemic infant may be hypertensive. Hypercalcemia also shortens the S-T segment and can lead to heart block and ultimately asystole. In older infants and young children with chronic hypercalcemia, poor growth and “failure to thrive” are often presenting manifestations. Hypercalcemia also leads to hypercalciuria, nephrocalcinosis, and nephrolithiasis. Hypercalcemia may also be present in patients with phenotypic abnormalities, for example, the Williams-Beuren syndrome (WBS).
Etiology
Causes of hypercalcemia in neonates and infants are listed in Tables 20.4A, 20.4B . Neonatal/infantile hypercalcemia may be iatrogenic in origin—for example, administration of excessive calcium or vitamin D—at times to the mother who secretes cholecalciferol in her breast milk; administration of thiazide diuretics that increase renal tubular absorption of calcium; or purposeful restriction of phosphate. Hypercalcemia, hypophosphatemia, hyperphosphatasemia, and radiographic evidence of rickets may develop in very premature infants receiving intravenous alimentation deficient in phosphate or in those fed only human breast milk, the phosphate content of which is low. Hypophosphatemia leads to increased synthesis of calcitriol by: (1) stimulating renal tubular 25OHD-1α hydroxylase activity directly, and (2) inhibiting synthesis of FGF23, thereby removing an agent that depresses activity of 25OHD-1α hydroxylase; the two pathways additively increase 25OHD-1α hydroxylase activity, calcitriol synthesis, and intestinal calcium absorption. The problem may be circumvented by increasing the amount of parenteral phosphate administered to the extent possible or by the use of breast milk fortified with phosphate. (Adequate extrauterine mineralization of the preterm skeleton requires intakes of both calcium and phosphate of approximately 200 mg/kg/d each.) Extracorporeal membrane oxygenation may also be associated with hypercalcemia in neonates, perhaps related to increased secretion of PTH by the neonate.
|
|
| Gene Protein Chromosome OMIM | Disorder OMIM | Clinical/Biochemical Manifestations | Gene Function/Transmission |
|---|---|---|---|
| CASR Calcium sensing receptor 3q13.3-21.1 601199 | Familial hypocalciuric hypercalcemia (FHH) type I, 145980; Neonatal severe hyperparathyroidism (NSHPT) 239200, Adult-onset primary hyperparathyroidism (AOPH) | Hypercalcemia, NSHPT—life-threatening AOPH—nephrolithiasis, low bone mineralization | Calcium receptor regulating synthesis, secretion of PTH, inactivating variant FHH1—AD NSHPT—AD/AR AOPH—AD/AR |
| GNA11 Guanine nucleotide-binding protein, alpha-11 19p13.3 139313 | Familial hypocalciuric hypercalcemia type II, 145981 | Hypercalcemia | Encodes G-protein transmitting signal of CaSR to intracellular signal transduction pathways, inactivating variant, AD |
| AP2S1 Adaptor related protein complex 2, sigma1 subunit 19q13.31 602242 | Familial hypocalciuric hypercalcemia type III, 600740 | Hypercalcemia, low bone mineralization, impaired cognition | Adaptor-related protein complex 2 interacts with β-arrestin ( ARRB1 , chr. 11q13.4, OMIM 107940) to form complex facilitating retrograde movement (endocytosis) of the CaSR from the cell surface & its return to the cytoplasm, inactivating variant AD |
| CYP24A1 Cytochrome P450, family 24, subfamily A, polypeptide 1 20q13.2 143880 | Infantile hypercalcemia 1 143880 | Hypercalcemia, vomiting, anorexia, inanition, nephrocalcinosis with inappropriate normal/high levels of calcitriol | Encodes 1,25-dihydroxyvitamin D-24-hydroxylase that inactivates calcitriol, AR |
| SLC34A1 Solute carrier family 34 (Type II sodium phosphate cotransporter), member 1, 5q35 182309 | Infantile hypercalcemia 2 616963 | Hypercalcemia, vomiting, anorexia, inanition, nephrocalcinosis | Encodes a proximal renal tubular sodium phosphate cotransporter, AR |
| TRPV6 Transient receptor potential, cation channel, subfamily V, member 6 7q34 606680 | Transient neonatal hyperparathyroidism 618188 | Skeletal demineralization in utero & postnatally, eucalcemia or hypocalcemia | Encodes a calcium selective channel active in the placental trophoblast, renal tubule, and intestinal tract, AR |
| Deletion 7q11.23 syndrome 194050 | Williams-Beuren syndrome: Infantile hypercalcemia | Hypercalcemia, supravalvular aortic stenosis, peripheral pulmonary arterial stenoses, characteristic face, developmentally challenged, friendly personality, verbal & musical skills | Deletion of 28 (±) contiguous genes including ELN |
| MEN1 Men1 gene 11q13.1 613733 | Multiple endocrine neoplasia, type 1 131100 | Tumors of the parathyroid, pancreatic islets, intestinal endocrine cells, adenohypophysis | Tumor suppressor, nuclear scaffold protein interacting with several transcription factors, inactivating variant AD |
| RET Rearranged during transfection protooncogene 10q11.21 164761 | Multiple endocrine neoplasia, type 2A 171400; type 2B 162300 | Medullary carcinoma of thyroid (2A,2B), pheochromocytoma (2A, 2B), parathyroid adenoma (2A), marfanoid habitus/ ganglioneuromas (2B) | Transmembrane receptor tyrosine kinase, activating variant, AD |
| CDKN1B Cyclin-dependent kinase inhibitor 1B 12p13.1 600778 | Multiple endocrine neoplasia, type 4 610755 | Parathyroid adenoma, gastric carcinoid, pituitary adenoma | Blocks cell cycle at G0/G1 phase, regulates cell motility, apoptosis, inactivating variant AD |
| CDC73 Cell division cycle protein 73, S cervisiae, homolog of 1q31.2 607391 | Familial isolated hyperparathyroidism-type 1 (145000) & type 2 (145001—associated with the hyperparathyroidism—jaw tumor syndrome) | Parathyroid adenoma/carcinoma, ossifying fibromas of mandible & maxilla, Wilms tumor | Parafibromin—protein intrinsic to the cell division cycle; tumor suppressor, inactivating variant, AD |
| GCM2 Glial cells missing, drosophila, homolog of, 2 6p24.2 603716 | Familial isolated hyperparathyroidism-type 4 617343 | Familial isolated hyperparathyroidism with multiple parathyroid adenomas, carcinoma | Encodes transcription factor essential for differentiation of the parathyroid glands, activating variant AD |
| PTH1R Parathyroid hormone receptor 1 3p21.31 168468 | Metaphyseal chondrodysplasia, Murk-Jansen type 156400 | Marked growth retardation, genu varum, small mandible, hypercalcemia, hypophosphatemia | G-protein coupled receptor for PTH & PTHrP, activating variant, AD |
Hypervitaminosis D may be caused by prolonged feeding of an improperly prepared formula or commercial dairy milk containing excessive vitamin D, iatrogenic prescription of vitamin D, calcidiol, or calcitriol, or increased endogenous production of calcitriol from inflammatory sites. In infants with severe birth trauma, perinatal asphyxia or hypothermia, subcutaneous fat necrosis may develop within the first 6 weeks after birth as manifested by indurated, extremely firm, tender, violaceous nodules on the cheeks, trunk, shoulders, buttocks, arms, and legs. Hypercalcemia may be present when the lesions first appear or develop as the nodules resolve several weeks later. Histologically, the skin lesions are composed of adipocytes, an inflammatory lymphohistiocytic infiltrate, and multinucleated giant cells in a bed of calcium crystals. The hypercalcemia of subcutaneous fat necrosis has been attributed to reabsorption of precipitated calcium, extrarenal synthesis of calcitriol by local macrophages and resultant hyperabsorption of ingested calcium, and increased osteoclastic activity because of enhanced prostaglandin secretion. The 25OHD-1α-hydroxylase activity of these inflammatory macrophages is not under the control of PTH, calcium, or phosphate, but is suppressible by glucocorticoids. Excessive production of prostaglandin E and interleukins (IL)-1 and -6 further contributes to hypercalcemia in this disorder by increasing the rate of bone turnover. Hypercalcemia attributable to subcutaneous fat necrosis is managed by the ingestion of a formula low in calcium, avoidance of vitamin D, and administration of fluids, calcitonin, or bisphosphonate (pamidronate), as needed. Administration of loop diuretics (e.g., furosemide) and glucocorticoids should be avoided if possible. Hypercalcemia caused by subcutaneous fat necrosis has also been observed in older children with major trauma or disseminated varicella. Congenital deficiency of lactase and other disaccharidases have also been associated with infantile hypercalcemia, likely because of increased intestinal absorption of calcium promoted by disaccharides.
Neonatal severe hyperparathyroidism (NSHPT) is a potentially lethal form of familial (hereditary) hypocalciuric hypercalcemia (HHC). It is most commonly caused by homozygous or compound heterozygous inactivating mutations of CASR that greatly increase the serum concentration of Ca 2 + needed to suppress PTH synthesis and secretion. However, in several infants with NSHPT, there have been heterozygous inactivating mutations of CASR (e.g., pArg185Gln, p.Arg227Leu) suggesting that the products of these mutations exert a dominant-negative effect on the normal CaSR, perhaps by interfering with migration of wild-type receptor to the cell surface or inactivation of wild-type receptor by linking to the mutated CaSR once embedded in the cell membrane, or by sequestration of G-proteins. In addition, the fetus bearing a heterozygous mutation in CASR that has been inherited from an affected father but residing in the womb of a normal mother may be relatively “hypocalcemic” in utero, leading to hyperplasia of the fetal parathyroid glands that persists after birth giving rise to NSHPT. Occasionally, NSHPT may be transmitted as an autosomal recessive disorder by clinically and biochemically normal parents. In subjects with homozygous mutations near the amino terminal of CASR (e.g., p.Leu13Pro), hypercalcemia may not be manifested until midchildhood or even adulthood. Thus the clinical spectrum of NSHPT ranges from mild (constipation, polyuria) with calcium concentrations ranging from 11 to 13 mg/dL (2.75–3.25 mmol/L) to severe and life-threatening (dysrhythmia, respiratory distress caused by hypotonia, demineralization and fractures of the ribs) when calcium levels exceed 15 mg/dL (3.75 mmol/L). Thus NSHPT may present within the first few days of life to several months of age depending on the degree of hypercalcemia. Search of the family history may identify members with mild hypercalcemia caused by autosomal dominant HHC. Physical examination often reveals hypotonia, feeding difficulties, and respiratory distress. The serum calcium concentration is frequently markedly elevated (average 14 mg/dL, range 11 to > 20 mg/dL; 3.5 mmol/L, 2.75 to 5 mmol/L) as is the PTH value (average 540 pg/mL, range 55 to > 1000 pg/mL). Other findings include: hypophosphatemia, hypermagnesemia, hyperphosphatasemia, and elevated calcitriol values, low renal tubular reabsorption of phosphate and relative hypocalciuria. Radiographically, there is evidence of hyperparathyroid bone disease—osteopenia, metaphyseal widening and irregularity, subperiosteal resorption, varus angulation of the hips, and fractures.
Lowering of the elevated serum calcium concentration present in NSHPT is only modestly affected by induction of diuresis by infusion of sodium chloride and by administration of furosemide; calcitonin and a bisphosphonate (e.g., pamidronate/zolendronate) may be required to suppress resorption of bone calcium and reduce serum calcium levels. These agents do not reduce the elevated level of PTH, however, and do not address the marked skeletal demineralization of hyperparathyroidism. The allosteric calcimimetic cinacalcet hydrochloride that acts directly on the CaSR has also been effective in lowering serum calcium concentrations in infants with NSHPT. (However, cinacalcet is not approved for use in subjects less than 18 years of age.) Subtotal parathyroidectomy may be a requisite lifesaving measure at times. Children with NSHPT who remain hypercalcemic are anorectic, fail to thrive, and at risk for developmental delay.
Secondary hyperparathyroidism in the neonate may be the result of maternal hypocalcemia caused by hypoparathyroidism, PHP, renal tubular acidosis, or vitamin D deficiency. Maternal hypocalcemia reduces placental transport and net delivery of calcium to the fetus resulting in relative fetal hypocalcemia, leading to hyperplasia of the fetal parathyroid glands and secondary hyperparathyroidism proportional to the maternal calcium deficit. Although only 25% of infants of hypocalcemic mothers are hypercalcemic, most have skeletal changes that reflect PTH excess that vary from severe demineralization with fractures to osteopenia detectable only by bone mineral densitometry. Secondary hyperparathyroidism usually resolves within a few weeks after birth as the infant ingests adequate calcium and phosphate. Normal transplacental maternal-fetal calcium transport is dependent upon three placental trophoblast processes: (1) apical Ca 2 + entry through Ca 2 + channels via an electrochemical gradient, (2) intracellular binding of Ca 2 + to calbindin D9k, and (3) extrusion of Ca 2 + through the basolateral membrane of the trophoblast. Transient neonatal hyperparathyroidism (OMIM 618188) may also occur in newborns with biallelic inactivating variants of TRPV6 (OMIM 606680) encoding a permeable calcium selective channel active in the placental trophoblast, renal tubule, and intestinal tract. In this disorder, decreased transplacental transport of calcium from the mother to the fetus leads to in utero fetal hypocalcemia and secondary hyperparathyroidism manifested by skeletal abnormalities, including thin, short bones, some with fractures that resolve within 2 years after birth. Mucolipidosis type II (OMIM 252500) has also been associated with neonatal secondary hyperparathyroidism; this Hurler-like disorder is characterized by facial abnormalities (asymmetry, flat nasal bridge), hepatosplenomegaly, skeletal deformities (dysostosis multiplex), and developmental delay and is caused by inactivating mutations in a gene ( GNPTAB , OMIM 607040) encoding N-acetyglucosamine-1-phosphotransferase, α/β subunits, a lysosomal phosphotransferase required for synthesis of mannose 6-phosphate. In this disease, maternal calcium concentrations are normal, but placental histology is abnormal suggesting impaired placental transport of calcium and fetal hypocalcemia, leading to compensatory increase in PTH generation by the fetus. In turn, skeletal evidence of PTH excess (osteopenia, fractures) develops; secondary hyperparathyroidism and its adverse effects remit within the first several weeks to months after birth. Transient neonatal distal renal tubular acidosis has also been associated with hypercalcemia because of secondary hyperparathyroidism. Occasionally, neonates with primary congenital hypothyroidism are transiently hypercalcemic.
Murk-Jansen metaphyseal chondrodysplasia (OMIM 156400) is an autosomal dominant chondrodystrophy associated with marked hypercalcemia as a consequence of heterozygous mutations that lead to constitutive, ligand-independent activation of PTH1R expressed in the kidney, bone, and growth plate chondrocytes. Enhanced functional PTH1R leads to accelerated chondrocyte proliferation, decreased rate of chondrocyte maturation, delayed bone formation, and increased bone resorption. Phenotypically, the patient with Murk-Jansen chondrodysplasia manifests short-limbed dwarfism; deformities of the long bones, digits, spine, and pelvis; choanal atresia; highly arched palate; micrognathia; widely open cranial sutures (in infancy); sclerosis of the basal cranial bones; disorganization of the metaphyses (delayed chondrocyte differentiation, irregularly calcified cartilage protruding into the diaphysis); and excessive loss of cortical bone but normal trabecular bone. Although birth length and physical appearance may be normal in affected neonates, there is radiographic evidence of the chondrodysplasia. Affected neonates are often eucalcemic, but later (infancy and childhood) develop hypercalcemia, hypophosphatemia, increased serum concentrations of calcitriol and elevated urinary excretion of nephrogenous cyclic AMP and calcium but low or undetectable serum levels of PTH and PTHrP. (Adults with this disorder are often eucalcemic, as well as very short [adult height 100 cm]; skeletal maturation is delayed; metaphyses of the long bones are long and histologically disorganized; and cranial bones are sclerotic.) Constitutively activating mutations of PTH1R in these subjects include most commonly p.His223Arg at the junction of the first intracellular loop and second transmembrane domain and p.Thr410Pro in the sixth transmembrane domain—sites that are specifically important in conferring ligand-independent activity upon PTH1R. SLK family kinase 3 ( SIK3 , OMIM 614776) is active in the PTH1R intracellular signal transduction pathway; a biallelic inactivating variant of SIK3 results in a hypercalcemic spondyloepimetaphyseal chondrodysplasia similar to but distinct from that of Murk-Jansen. Excessive secretion of PTHrP and resultant hypercalcemia have also been recorded in infants with neonatal iron storage disease and embryonal renal tumors (Wilms, mesoblastic nephroma).
WBS (OMIM 194050) is a hemizygous contiguous gene deletion syndrome (involving approximately 26–28 genes on chromosome 7q11.23) that is transmitted as an autosomal dominant disorder and whose prevalence is 1/7500 to 1:15,000 live births. The WBS is characterized by intrauterine and postnatal growth retardation, hypercalcemia in infancy in 15% of patients that usually resolves by 2 years of age but may occasionally persist into adulthood, consistent hypercalciuria, supravalvular aortic stenosis in 30% of subjects, narrowing and stenoses of the thoracic aorta and coronary, pulmonary, renal, mesenteric, and celiac arteries, hypertension, microcephaly, “elfin” face (broad forehead, epicanthal folds, stellate iris pattern, esotropia, short nose with full nasal tip, arched upper and prominent lower lips, long philtrum, full cheeks with flattened malar eminences, dental malocclusion), hoarse voice, hyperacusis in childhood associated with nerve deafness, radioulnar synostosis, renal hypoplasia or unilateral agenesis, developmental delay (poor visual-motor integration, attention deficit disorder, mean IQ 58, range 20–106), and short stature. Although most patients with WBS are developmentally challenged, they have unique and proficient verbal skills with a large vocabulary and enhanced auditory memory particularly for names, adept social language skills, and exceptional musical aptitude including the ability to memorize and sing many musical compositions and play many instruments. Children with WBS prefer to be in the company of adults rather than with their peers and are not shy or fearful of strangers.
Hypercalcemia in subjects with the WBS may be pathogenetically related to partial loss of function of the Williams syndrome transcription factor (WSTF) encoded by BAZ1B (Bromodomain adjacent to zinc-finger domain, 1B, OMIM 605681). The WSTF is a nuclear protein that is part of a multimeric, ATP-dependent, chromatin remodeling complex termed WINAC (WSTF including nucleoside assembly complex). Independently of ligand-binding, the VDR interacts with WINAC. Under usual circumstances, binding of calcitriol to the VDR-WINAC complex (1) represses expression of renal tubular CYP27B1 , the gene encoding 25-hydroxyvitamin D-1α-hydroxylase, and (2) enhances transcription of CYP24A1 , the gene encoding 25-hydroxyvitamin D-24-hydroxylase. Hence haploinsufficiency of WSTF may lead to increased and relatively unregulated synthesis of calcitriol while slowing its rate of degradation; in response to the increase in calcitriol generation, the intestinal absorption of calcium increases and hypercalcemia ensues. Other endocrinopathies associated with WBS include glucose intolerance and hypothyroidism.
WBS is the consequence of deletion of chromosome 7q11.23 caused by unequal meiotic recombination (unequal crossover of genes between chromosome 7 homologs during meiosis); this genetic error occurs with equal frequency in the gametes of both parents and results in the loss of 26 to 28 genes. Hemizygosity of ELN (OMIM 130160) encoding elastin is the attributed cause of the cardiovascular malformations and hypertension observed in WBS, inasmuch as partial loss of ELN leads to compensatory increase in the number of rings of smooth muscle and elastic lamellae resulting in arterial thickening and increased risk of obstruction. Functional hemizygosity for general transcription factor II-I ( GTF2I , OMIM 601679), GTF2IRD1 (OMIM 604238), HIP1 (OMIM 601767), and YWHAG (OMIM 605356) may underlie the neurocognitive deficits and strengths that characterize WBS and their distinctive personalities. Haploinsufficiency of LIM domain kinase 1 ( LIMK1 , OMIM 601329) may also be linked to the neurocognitive defects in patients with WBS. Hemizygous loss of STX1A (OMIM 186590) may be pathogenetically related to development of glucose intolerance in WBS subjects. The WBS phenotype has also been associated with interstitial deletion of chromosome 6q22.2-q23, as well as defects in chromosomes 4, 11, and 22—implying that the syndrome is genetically heterogeneous. The diagnosis of the WBS syndrome is suspected on the basis of the characteristic clinical phenotype (with/without hypercalcemia) and confirmed by demonstration of the microdeletion at chromosome 7q11.23 by microarray or FISH, although a normal chromosome analysis does not entirely eliminate this diagnosis. Specific genotyping may also prove diagnostically useful on occasion. Hypercalcemia is managed by ingestion of a low calcium, vitamin D-free formula; occasionally short-term glucocorticoid therapy may be necessary to restore eucalcemia.
A contiguous gene deletion syndrome characterized by developmental delay and seizures involving a segment of chromosome 7q11.23 distal to the deletion associated with the WBS has been described (OMIM 613729). Duplication of chromosome 7q11.23 results in a syndrome associated with heart defects, diaphragmatic hernia, cryptorchidism, and neurocognitive and behavioral aberrations. Additional findings in patients with dup7q11.23 include short stature and minor dysmorphic facial features (prominent forehead, long nasal tip, short philtrum, thin lips, retrognathia, folded ear helices). Neurocognitively, there is severe speech delay and often autistic behavior but intact visuocognitive skills—characteristics that are opposite to those present in patients with the WBS.
Although phenotypically normal, infants with hypercalcemia of infancy types 1 and 2 manifest anorexia, impaired growth, developmental delay, febrile episodes, polyuria, and vomiting, leading to dehydration, hypercalciuria, and nephrocalcinosis associated with hypercalcemia in the presence of decreased serum concentrations of PTH. Infantile hypercalcemia type 1 (OMIM 143880) is characterized by elevated or inappropriately normal levels (relative to serum calcium concentrations) of calcitriol most often caused by biallelic inactivating variants of CYP24A1 (OMIM 126065) encoding 24-hydroxylase, the enzyme that converts 25-hydroxyvitamin D to 24,25 dihydroxyvitamin D and 1,25-dihydroxyvitamin D to 1,24,25-trihydroxyvitamin D, thereby increasing their solubility and subsequent excretion in bile and urine. Delaying the degradation of 1,25-dihydroxyvitamin D prolongs its effective biological life. In many patients with infantile hypercalcemia type I, hypercalcemia resolves within the first several years of life but it may persist to older ages. Inactivating mutations in CYP24A1 have also been found in adults with hypercalcemia, hypercalciuria, and nephrolithiasis. CYP24A1 mutations may either decrease the binding of the enzyme to its substrate or interfere with the interaction of heme and enzyme protein; either type of mutation decreases its function. Avoidance of vitamin D, low dietary calcium intake, increased fluids, and occasionally glucocorticoids to reduce intestinal absorption of calcium are therapeutic modalities for this disorder. Infantile hypercalcemia type 2 (OMIM 616963) is the result of biallelic variations in SLC34A1 encoding solute carrier family 34 (Type II sodium phosphate cotransporter), member 1 (OMIM 182309), a proximal renal tubular sodium phosphate cotransporter, and is manifested by vomiting, anorexia, inanition, hypercalcemia, hypophosphatemia, hypercalciuria, and nephrocalcinosis. In this disorder, decreased renal tubular transport of phosphate results in hyperphosphaturia and hypophosphatemia, leading to decreased synthesis of FGF-23 and consequently to increase in functional activity of 25OHD-1α hydroxylase ( CYP27B1 ) and the ensuing synthesis of calcitriol and hyperabsorption of intestinal calcium. Restricted intake of calcium and vitamin D have also been used in the management of patients with infantile hypercalcemia type 2, but phosphate supplementation may also be considered. As adults, patients with variants of either CYP24A1 or SLC34A1 are of normal height and weight and in reasonably good health, although in some subjects, nephrocalcinosis persists. Some affected patients, however, develop progressive renal failure as adults.
Antenatal/neonatal Bartter syndromes type 1 (OMIM 601678) and type 2 (OMIM 241200) are clinically and biochemically similar and are caused by biallelic loss-of-function mutations in genes controlling a transepithelial sodium-potassium-chloride transporter ( SLC12A1 , OMIM 600839) and an inwardly rectifying apical potassium channel ( KCNJ1 , OMIM 600359), respectively, expressed in the renal tubular TALH. Affected fetuses develop polyhydramnios, leading to premature delivery with postnatal salt-wasting and secondary hyperaldosteronism; increase renal and systemic production of prostaglandin E2 inhibits sodium and chloride reabsorption in the TALH and enhances juxtaglomerular renin release resulting in hypokalemic metabolic acidosis, hyperparathyroidism, hypercalcemia, hypercalciuria, nephrocalcinosis, hypomagnesemia, osteopenia, and often death in type 1 antenatal Bartter syndrome. Type 2 neonatal Bartter syndrome caused by variants of KCNJ1 is manifested by hypokalemia (occasionally transient hyperkalemia), reduced intravascular volume, and increased levels of angiotensin leading to renal and systemic production of prostaglandin E2 that also impair renal tubular reabsorption of sodium and chloride in the TALH. Hypochloremic, hypokalemic alkalosis, hyperprostaglandin E, hypercalcemia, hypercalciuria, and nephrocalcinosis suggest neonatal Bartter syndrome. Replacement of fluid and electrolytes and administration of potassium-sparing diuretics (thiazides) and cyclooxygenase inhibitors (indomethacin) may be effective in ameliorating the biochemical and clinical manifestations of the disease.
Neonatal or infantile hypercalcemia may occur in patients with one of several inborn errors of metabolism. Both congenital lactase deficiency (OMIM 223000) caused by mutations in LCT (OMIM 603202) and sucrase-isomaltase deficiency (OMIM 222900) caused by variants of SI (OMIM 609845) have been associated with infantile hypercalcemia, likely caused by increased intestinal absorption of calcium promoted by dissaccharides. Hypercalcemia may also be encountered in infants with primary oxalosis (OMIM 259900) that may be the result of variants in one of three genes ( AGXT , OMIM 604285; GRHPR , OMIM 604296; DHDPSL , OMIM 613597). The “blue diaper syndrome” (OMIM 211000) is the result of inability of the gastrointestinal tract to absorb tryptophan that intestinal bacteria metabolize to an indole that is absorbed and excreted in the urine resulting in indicanuria that is then oxidized to the pigment “indigo blue” thus staining the diaper of the affected neonate and infant. Hypercalcemia, hypercalciuria, and nephrocalcinosis are common but not invariate; the pathogenesis of hypercalcemia in these patients is unknown. This disorder has been associated with a homozygous loss-of-function mutation in PCSK1 encoding a prohormone convertase/calcium-dependent serine endoproteinase (proprotein convertase, subtilisin/kexin-type 1, OMIM 162150) that converts a multicomponent protein to specific bioactive protein products. Variants of PCSK1 have also been related to defects in insulin synthesis, as well as several endocrinopathies (hypogonadotropism, central hypoadrenocorticism, and central diabetes insipidus). Hypercalcemia may also occur in patients with the IMAGe syndrome of intrauterine growth retardation, metaphyseal dysplasia, adrenal hypoplasia congenita, and genital anomalies (OMIM 614732) caused by heterozygous variants of CDKN1C encoding cyclin-dependent kinase inhibitor 1C (OMIM 600586), a regulator of early cell division.
Hypophosphatasia (HPP) is a congenital disorder of bone mineralization resulting in rickets of variable severity that is caused by a decreased osteoblast synthesis of tissue nonspecific alkaline phosphatase (TNSALP)—the consequence of monoallelic or biallelic inactivating mutations in ALPL (OMIM 171760) (vide infra). Decreased production of alkaline phosphatase leads to a deficit in phosphate ions impairing synthesis of hydroxyapatite resulting in rickets, whereas continued intestinal absorption of calcium can lead to hypercalcemia. The infantile form of hypophosphatasia (OMIM 241500) is caused by biallelic loss-of-function variants of ALPL . It becomes clinically apparent after the first month of life but usually before 6 months of age and is often fatal. Initial symptoms are failure to thrive associated with impaired feeding, weakness, and delayed achievement of motor milestones; rachitic deformations of the thorax and limbs are visible. Radiographically, it is characterized by demineralization of the calvarium and peripheral skeleton, rickets, spurs of cartilage and bone extending from the sides of the knee and elbow joints, and premature craniosynostosis, leading to increased intracranial pressure. Also present are vitamin B 6 -dependent seizures, hypercalcemia, and hypercalciuria. Defective osteoblast synthesis of TNSALP is the result of loss-of-function missense, nonsense, and donor splice site mutations of ALPL . The lethal homozygous or compound heterozygous mutations of ALPL are located within or near the enzyme domain and/or the homodimer and tetramer interfaces. Inappropriately low serum bone alkaline phosphatase activity differentiates this illness from other rachitic or osteopenic states (osteogenesis imperfecta congenita [OIC] or achondrogenesis type 1A) in which alkaline phosphatase activity is usually normal or elevated. Increased urine phosphoethanolamine and serum inorganic pyrophosphate (an inhibitor of hydroxyapatite crystallization that also impairs skeletal mineralization) and pyridoxal-5′-phosphate values are consistent with the diagnosis of hypophosphatasia, whereas analysis of ALPL identifies the gene mutation(s) itself. The diagnosis of hypophosphatasia can be suspected on prenatal ultrasonography and confirmed by ALPL genotyping. The hypercalcemia of infantile hypophosphatasia is managed by hydration, diuretics that act upon the TALH (e.g., furosemide), and administration of bisphosphonates (pamidronate), calcitonin, or glucocorticoids as necessary. Dietary calcium intake should be restricted and vitamin D and its metabolites avoided. Bone marrow transplantation and “stem cell boosts” of transfused donor osteoblasts have also been used to treat affected patients. Administration of a bone-targeted form of TNSALP has been used successfully to treat life-threatening hypophosphatasia and is currently FDA approved for this purpose.
Hypercalcemia and hypercalciuria of unknown pathogenesis occur occasionally in subjects with the IMAGe syndrome (OMIM 614732) caused by gain-of-function variants of CDKN1C (OMIM 600856, chr. 11p15.4) encoding a paternally imprinted (maternally transmitted) cyclin-dependent kinase inhibitor critical for normal regulation of the cell’s mitotic processes. Craniosynostosis, cleft palate, and scoliosis may also develop in these patients.
Evaluation and Management
After historical review (the family history is explored for members with mineral disorders and maternal status considered in depth, and the infant’s intake of calcium, phosphate, and vitamin D is estimated) and physical examination (nonspecific manifestations of hypercalcemia include—hypertension, subnormal growth, hypotonia, weakness, lethargy, stupor, occasionally seizures; specific findings associated with hypercalcemia include—facial and cardiovascular signs of WBS, subcutaneous nodules consistent with fat necrosis, skeletal deformities suggestive of metaphyseal chondrodysplasia or infantile hypophosphatasia) have been completed, evaluation of the hypercalcemic neonate and infant continues with measurements of total serum calcium and Ca 2 + , phosphate, alkaline phosphatase, PTH, calcidiol, and calcitriol and determination of urine calcium and creatinine levels ( Fig. 20.5 ). Hypocalciuria in the presence of hypercalcemia suggests the presence of NSHPT. Occasionally, one may encounter a neonate in whom the total calcium concentration is elevated because of hyperproteinemia but the Ca 2 + value is normal, a state termed pseudohypercalcemia . In infants with suspected WBS, microarray or FISH analysis searching for deletion of chromosome 7q11.23 should be undertaken. In those in whom the diagnosis of NSHPT is being considered, determination of parental serum and urine calcium and creatinine values is often helpful unless the disorder has been transmitted as an autosomal recessive disease. Genotyping of CASR should be undertaken when NSHPT is suspected.
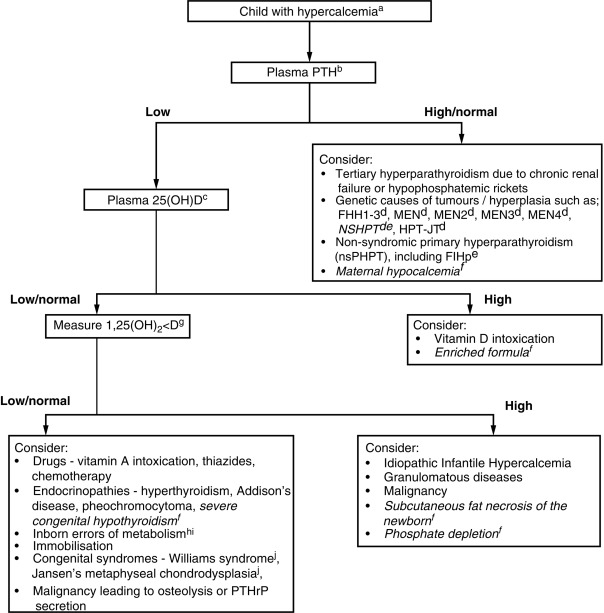
Treatment of hypercalcemia in neonates and infants must be directed to prevention of progression, identification of cause, and assessment of severity. Use of a formula low in calcium and avoidance of vitamin D (excessive intake or sunlight) are helpful in the majority of neonates with modest hypercalcemia. Infants with significantly elevated serum calcium levels, particularly if long-standing, are often dehydrated. Immediate treatment consists of infusion of 0.9% sodium chloride with 30 mEq/L of potassium chloride (10–20 mL/kg over 1 hour); after dehydration has been corrected and adequate urine flow established and if substantial hypercalcemia persists an intravenous bolus injection of furosemide (1–2 mg/kg) may be administered. Currently, bisphosphonates, analogs of pyrophosphate that adhere to the surface of hydroxyapatite crystals and are incorporated by osteoclasts thereby inhibiting osteoclast-mediated dissolution of bone mineral and matrix, are the agents of choice for the treatment of substantial hypercalcemia in infants. Pamidronate (0.5–2.0 mg/kg in 30 mL normal saline intravenously over 4 hours) has been successfully used in infants with hypercalcemia caused by vitamin D intoxication, subcutaneous fat necrosis, NSHPT, and other causes. The effects of bisphosphonate last for several weeks to months and when administered in excess can lead to hypocalcemia, hypophosphatemia, and hypomagnesemia. Other agents that may be considered in the treatment of a hypercalcemic infant include salmon calcitonin (2–4 units/kg subcutaneously every 6–12 hours) to inhibit calcium mobilization from bone. Although the type II calcimimetic—cinacalcet—has been demonstrated to improve function of many inactive mutant forms of the CASR and to be of clinical benefit in adults with familial hypocalciuric hypocalcemia, this agent is not approved for use in children in the United States, a therapeutic trial of this agent may be considered in a patient with NSHPT with appropriate safeguards and permissions. In the newborn with life-threatening NSHPT, total parathyroidectomy may be urgently required. Postoperatively, the patients may become hypocalcemic as calcium is avidly deposited in bone (hungry bone syndrome) and require large amounts of supplemental calcium until stabilized.
Infants with hypercalcemia caused by WBS (that often remits by 2 years of age) or with some forms of osteopetrosis may be managed by use of a low-calcium formula (e.g., Calcilo XD—2.9 mg of calcium per 100 calories; free of vitamin D) and withholding of supplemental vitamin D. Infants with “idiopathic” hypercalcemia of infancy caused by inactivating mutations of CYP24A1 should receive the low-calcium formula and supplemental vitamin D should be withheld. Some children with hypercalcemia caused by inactivating mutations of CYP24A1 respond to ketoconazole, which inhibits conversion of calcidiol to calcitriol or rifampin, which also inactivates metabolites of vitamin D. Patients with mutations in SLC34A1 may respond to administration of supplemental phosphate. Mineral homeostasis must be monitored closely in infants receiving a low-calcium formula and from whom vitamin D is being withheld to prevent deficiencies of both nutrients and development of secondary hyperparathyroidism.
Hypercalcemia in the Child and Adolescent
Hypercalcemia is present in the child/adolescent when the serum calcium concentration exceeds plus two standard deviations above the normal mean; for subjects 4 to 18 years of age these values are: total calcium 10.7 mg/dL (2.70 mmol/L); Ca 2 + 5.52 mg/dL (1.38 mmol/L). Hypercalcemia is most often asymptomatic in the older child and adolescent and identified unexpectedly by routine serum chemical analysis for an unrelated problem. Clinical manifestations of hypercalcemia in the child/adolescent include: anorexia, nausea, vomiting, constipation, abdominal or flank pain, polydipsia, polyuria, renal colic, altered alertness, lethargy, hypotonia, irritability, or seizures. Unrecognized, chronic hypercalcemia may impair growth, lead to nephrocalcinosis and nephrolithiasis preceding renal failure, pancreatitis, impaired cognition, or symptoms of mental illness. Long bone fracture through a fibrous cystic “brown tumor” (osteitis fibrosa cystica) may be a presenting complaint. Physical findings are nonspecific and may include hypertension, weakness, hyporeflexia, dull affect, aberrant mentation, or flank tenderness.
Etiology and Pathogenesis
Causes of hypercalcemia in children and adolescents are listed in Tables 20.4A, 20.4B . In the presence of a normal serum protein concentration, hypercalcemia occurs when the “set point” for serum Ca 2 + is increased because of a loss-of-function mutation in CASR or the set point is reversibly increased by lithium, or when the rate of entry of calcium into the extracellular and circulatory compartments from bone, intestinal tract, or kidney exceeds its rate of loss. The resorption rate of bone mineral may be increased by excessive secretion of PTH or PTHrP, constitutive activation of PTH1R, excessive intake of vitamin D or metabolites, increased production of osteoclast-activating inflammatory cytokines or localized osteolytic processes, such as metastatic neoplasms, and dissociation of the rates of bone formation and resorption—for example, immobilization. The absorption rate of intestinal calcium may be increased by excessive intake of calcium, by hypervitaminosis D of exogenous or endogenous origin, or by increased secretion of PTH or PTHrP. Augmented renal tubular reabsorption of filtered calcium may occur with administration of calcium-sparing diuretics, such as thiazides, an effect that may “unmask” hypercalcemia in a previously eucalcemic patient with hyperparathyroidism. Thus hypercalcemia may be considered to be PTH mediated or PTH independent. In the presence of hyperalbuminemia, the total serum calcium concentration but not the Ca 2 + concentration is increased (pseudohypercalcemia). Venous stasis (e.g., by tourniquet during blood sampling) results in spuriously altered local pH and Ca 2 + values.
Hyperparathyroidism is most often caused by a single parathyroid adenoma but may also be caused by hyperplasia of all four parathyroid glands as in patients with HHC caused by loss-of-function variants of CASR , GNA11 , or AP2S1 . Respectively, these genes encode the plasma membrane heptahelical CaSR, its guanosine triphosphate–coupled signal transduction factor (Gq/11α), and an adaptor-related protein complex that facilitates endocytosis of the CaSR. Familial HHC type 1 (HHC1, OMIM 145980) is an autosomal dominant disorder with complete penetrance at all ages characterized by PTH-dependent, usually asymptomatic (total and ionized) hypercalcemia with hypocalciuria (and, therefore absence of nephrocalcinosis or calcium-containing renal calculi), hypermagnesemia, hypomagnesuria, and hypophosphatemia that is caused by heterozygous loss-of-function mutations in CASR (OMIM 601199). Consequently, higher serum concentrations of Ca 2 + are required to activate the CaSR and inhibit synthesis and secretion of PTH. Serum concentrations of PTH may be normal or slightly elevated, but inappropriately high for the Ca 2 + level; calcidiol and calcitriol values are normal. In children, HHC1 is most commonly suspected initially by the presence of unexpected hypercalcemia (11–13 mg/dL) in a chemistry profile or through family screening of a parent or other relative with hypercalcemia. Although patients with HHC1 are hypercalcemic, they are paradoxically hypocalciuric because inactivation of the CaSR results in enhanced distal renal tubular reabsorption of filtered calcium. PTH levels are normal or slightly elevated. When mutations in CASR are biallelic or when a neonate with a monoallelic variant of CASR is born to a normocalcemic woman, hypercalcemia may become extreme and even life-threatening (NSHPT). Older subjects with HHC1 may complain of fatigue, weakness, or polyuria; there is a slightly increased incidence of relapsing pancreatitis, cholelithiasis, chondrocalcinosis, and premature vascular calcification in subjects with HHC1, but bone mass and fracture rate are normal. Because of decrease in the number or functional competence of parathyroid chief cell membrane CaSRs, the set point for Ca 2 + suppression of PTH secretion is reset upward. The parathyroid glands are slightly hyperplastic. In renal tubular cells, decrease in the number and activity of CaSRs results in increased renal tubular reabsorption of filtered calcium and relative hypocalciuria (ratio of calcium clearance/creatinine clearance < 0.01 in 80% of patients with HHC1); because the renal tubular reabsorption of magnesium is also increased, hypermagnesemia and hypomagnesuria are present; urinary concentrating ability and other measures of renal function are normal; in hypercalcemia of other pathogenesis, urinary calcium excretion is increased and renal concentrating function depressed. Usually, HHC1 requires no therapy, but must be differentiated from mild primary hyperparathyroidism in which hypomagnesemia and hypercalciuria are present. Subtotal parathyroidectomy in HHC1 does not lower calcium levels as the residual parathyroid glands hypertrophy; total parathyroidectomy is unnecessary except in a rare infant with NSHPT. More than 100 nonsense, missense, insertion, and deletion mutations in CASR associated with HHC1 or NSHPT have been identified, mostly in the receptor’s extracellular Ca 2 + binding domain; these mutations either decrease receptor affinity for Ca 2 + or alter intracellular processing of the CaSR (glycosylation, dimerization—e.g., p.Arg66His, p.Asn583Stop) in the endoplasmic reticulum preventing its translocation to the surface of the cell membrane ( Fig. 20.6 ). Many of the mutations are located in the extracellular domain of CaSR between codons 39 to 300, a region rich in aspartate and glutamate residues in which Ca 2 + may nestle. Glycosylation is necessary for both dimerization and trafficking of the CaSR. Missense mutations involving arginine at codon 66 (p.Arg66His/Cys) result in a product that is able to be only partially glycosylated; it can form homodimers in the endoplasmic reticulum but cannot enter the Golgi apparatus and be transported to the cell’s plasma membrane. Experimentally, approximately 50% of the products of inactivating mutations of CASR can be “escorted” to the plasma membrane and functionally stabilized by the allosteric calcimimetic cinacalcet proffering a potential therapeutic agent for the management of patients with symptomatic hypercalcemia because of loss-of-function CASR mutations, especially those with NSHPT, if it proves safe to do so in children. Mutations in CASR tend to be unique to the affected family. In approximately one-third of families with HHC1, no mutations in the coding region of CASR have been identified; thus they may have a mutation in a noncoding region of CASR or a variant of GNA11 or AP2S1 . Although the phenotypes are similar, these are two other genetic causes of hypocalcemic hypercalciuria designated types 2 and 3. Hereditary hypocalcemic hypercalciuria type 2 (OMIM 145981) is caused by loss-of-function variants of GNA11 (OMIM 139313), encoding a CaSR G-protein transmitting signal (Gα11) to intracellular signal transduction pathways. Hereditary HHC type 3 (OMIM 600740) is the consequence of inactivating mutations in AP2S1 (OMIM 602242) encoding a protein complex that interacts with β-arrestin ( ARRB1 , OMIM 107940, chr. 11q13.4) forming a structure enabling movement of the CaSR from the cell membrane and its return to the cytoplasm; as adults, patients with HHC3 may have hypophosphatemic osteomalacia. Autoantibodies against the amino terminal extracellular domain of the CaSR that reversibly inhibit receptor activity have been identified in patients with acquired HHC; this disorder may be responsive to glucocorticoid therapy. In some patients with primary or uremic secondary hyperparathyroidism, there is reduced expression of CASR and an increased set point for suppression of PTH secretion.

Primary hyperparathyroidism is an unusual childhood disorder with an incidence of 2 to 5/100,000 as compared with that in adults of approximately 100/100,000. In adults with hyperparathyroidism, females outnumber males 3:1; in children the female/male ratio is closer to one. In older children and adolescents, primary hyperparathyroidism is most often a sporadic disease and usually the result of a single parathyroid adenoma. Hyperparathyroidism may occur as an autosomal dominant disorder in patients with familial isolated primary hyperparathyroidism involving variants of CDC73 (OMIM 607391) and GCM2 (OMIM 603716) (vide infra). Familial hyperparathyroidism also occurs in patients with variants of the genes responsible for the syndromes of multiple endocrine neoplasia (MEN) type 1 ( MEN1 , OMIM 613733), MEN types 2/3 ( RET , OMIM 164761), and MEN type 4 ( CDKN1B , OMIM 600778).
MEN types 1 and 4 are clinically similar in that both entities are associated with neoplastic lesions of the parathyroid glands, adenohypophysis (somatotropinoma, prolactinoma), and endocrine secreting cells of the intestinal (gastrinoma, insulinoma) or genitourinary tracts. MEN1 is the consequence of heterozygous loss-of-function mutations in MEN1 encoding a nuclear scaffold protein that interacts with multiple transcription factors and is a tumor suppressive agent. MEN2A is associated with loss-of-function variants of RET encoding a transmembrane receptor tyrosine kinase that is associated with parathyroid adenoma, medullary carcinoma of the thyroid, and pheochromocytoma; RET variants associated with MEN2A involve cysteine residues in its extracellular domain. MEN2B is associated with medullary carcinoma of the thyroid (MCT) that is highly aggressive, pheochromocytoma, thickening of corneal nerves, mucosal ganglioneuromas on the tongue, lips, and eyelids, and a marfanoid habitus; genetically, MEN2B is related to the RET variant p.Met918Thr in exon 16, the site of the receptor’s tyrosine kinase domain. MEN4 is associated with adenomas of the parathyroid gland, adenohypophysis, and gastrointestinal tract that is the consequence of monoallelic inactivating variants of CDKN1B , a cyclin-dependent kinase inhibitor that blocks the cell cycle at the G0/G1 phase. (Cyclins are cell proteins that regulate progression of the cell cycle of growth, division, and duplication by activating selected cell cycle–related kinases [phosphorylases].) Familial isolated hyperparathyroidism type 1 (OMIM 145000) and familial isolated hyperparathyroidism type 2 associated with fibrous tumors of the maxilla and mandible (OMIM 145001) are caused by loss-of-function mutations in CDC73 (OMIM 607391) encoding a tumor suppressor. Familial isolated hyperparathyroidism type 3 (610071) has been mapped to chromosome 2p14-p13.3. GCM2 (OMIM 603716) encodes a transcription factor essential for differentiation of the parathyroid glands. Gain-of-function variants of GCM2 result in familial isolated hyperparathyroidism type 4 (OMIM 239200) with development of multiple parathyroid adenomas and occasionally carcinomas. NSHPT (OMIM 239200) is the consequence of inactivating mutations of CASR (OMIM 601199).
Children and adolescents with hypercalcemia caused by hyperparathyroidism may be asymptomatic or may display personality and behavioral changes—particularly depression, headache, malaise, proximal muscle weakness, anorexia, abdominal cramping, nausea and vomiting, constipation, polydipsia and polyuria or have symptoms reflecting the consequences of this disorder, such as flank pain and hematuria caused by renal calculi (hypercalciuria), abdominal pain (pancreatitis), or pathological fractures (through areas of osteopenia or lesions of osteitis fibrosa cystica). Rarely is it possible to palpate a cervical mass in these patients, although the physical examination may reveal slight weakness of proximal musculature. Hypercalcemia, hypophosphatemia, and elevated serum concentrations of intact PTH are present in the majority of children with hyperparathyroidism. In subjects with primary hyperparathyroidism, hypercalcemia is the result of increased secretion of PTH caused by loss of the normal relationship between the set point of serum Ca 2 + and PTH synthesis and release and to Ca 2 + -independent (constitutive) PTH secretion related to the mass of parathyroid tissue. Ultrasonography, magnetic resonance imaging, computed tomography (CT), and radionuclide scans ( 99m technetium-labeled sestamibi-single photon emission CT scan) have been used to localize the abnormal parathyroid gland(s) before surgical excision. (Sestamibi is methoxyisobutylisonitrile, a lipophilic cation.) Occasionally, the parathyroid tumor may be located ectopically in the thymus, thyroid gland, or mediastinum. Further studies may reveal subperiosteal bone resorption, nephrocalcinosis, or nephrolithiasis.
Pathologically, hyperparathyroidism in children is most often because of a chief cell adenoma involving one parathyroid gland, but adenomas in several parathyroid glands and diffuse hyperplasia (particularly in patients with MEN type 1), and rarely carcinoma of the chief cells may also occur. The majority of parathyroid adenomas are monoclonal in origin; that is, a single mutant cell develops into a tumor. In adolescents, parathyroid tumors may occasionally develop after external radiation of the neck for treatment of lymphoma. In some parathyroid tumors, increased expression of cyclin D1 (encoded by CCND1 , OMIM 168461, chr. 11q13.3) has been demonstrated. Overexpression of CCND1 in parathyroid chief cells is at times the result of a somatic chromosome mutation—inversion (rotation) of regions 11p15 and 11q13 in which the promoter region of PTH is repositioned to serve as a promoter for CCND1 , thereby increasing the rate of chief cell division whenever the (hypocalcemic) stimulus for PTH generation is received and leading ultimately to (benign) tumor formation. However, in many parathyroid adenomas, there is increased activity of cyclin D1 without this chromosomal rearrangement. In patients with parathyroid adenomas and carcinomas, overexpression of the retinoblastoma and p53 tumor-suppressor genes, whose products normally inhibit the cell cycle at the G1/S step, has also been demonstrated as have somatic mutations in CDC73. In approximately 35% of sporadic parathyroid adenomas, a somatic loss-of-function mutation of the tumor suppressor factor menin—the germline mutation in patients with MEN type 1—can be identified together with loss of heterozygosity on chromosome 11. Germline loss-of-function mutations in MEN1 and CASR and gain-of-function variants in GCM2 have also been detected in patients with isolated primary hyperparathyroidism in association with multiglandular involvement; in this instance, patients with CASR mutations may not have the typical biochemical findings of HHC1 (see Table 20.4B ). Chronic renal insufficiency leads to secondary hyperparathyroidism because of hyperplasia of the parathyroid glands and also to monoclonal parathyroid tumors (tertiary hyperparathyroidism) associated with somatic chromosomal deletions in some instances.
Familial isolated hyperparathyroidism type 1 (OMIM 145000) and type 2 (OMIM 145001—hyperparathyroidism associated with familial benign and malignant parathyroid tumors or multiple ossifying fibromas of the maxilla and mandible, respectively) are both caused by germline mutations in CDC73 (OMIM 607393, chr. 1q31.2) encoding parafibromin, a protein intrinsic to the cell division cycle. Hyperparathyroidism occurs in 80% of subjects with a mutation in CCD73 at a mean age of 32 years but may also appear in children before 10 years of age. In these patients, the parathyroid lesion may be an atypical, potentially premalignant cystic adenoma (65%), hyperplasia (20%), or even carcinoma (15%); the parathyroid tumor may be an isolated finding or it may be associated with maxillary and/or mandibular bone tumors composed of ossified fibrous tissue; renal (Wilms tumor, papillary renal cell carcinoma, hamartoma, polycystic kidney), pancreatic (carcinoma), and uterine (tumor) lesions may also develop in these patients. Lesions within the parathyroid glands may develop asynchronously. CDC73 is a 17 exon gene that encodes parafibromin, a 531 aa nuclear protein that is a component of a complex of accessory factors that modulates the activity of RNA polymerase II and of a histone methyltransferase and hence regulates gene expression and cell proliferation—functioning as both a transcriptional activator and repressor. For a parathyroid tumor to develop, a “second hit” must occur that results in loss of heterozygosity: that is, the germline inactivating mutation of CDC73 on one allele must be matched by a mutation in or deletion of CDC73 in the remaining normal allele. When a germline mutation in CDC73 has been detected, screening of family members for this mutation and longitudinal evaluation of affected subjects is recommended as asymptomatic individuals with atypical adenoma or carcinoma of the parathyroid glands may be so identified. Somatic mutations in CDC73 have also been identified in atypical parathyroid adenomas and many parathyroid carcinomas but are unusual in the typical sporadic parathyroid adenoma. Familial isolated hyperparathyroidism type 3 (OMIM 610071) has been linked to chromosome 2p14-13.3 but a specific gene variant in this site has not been identified. Hyperparathyroidism type 4 (OMIM 617343) is the result of variants of GCM2 (OMIM 603716) whose product is essential for differentiation of the parathyroid glands.
The syndromes of MEN are familial autosomal dominant diseases of high penetrance associated with the development of tumors in two or more endocrine glands within a single individual. There are four defined MEN syndromes—1, 2A, 2B, and 4 ( Table 20.5 ). MEN1 is characterized by development of tumors of the parathyroid glands (adenoma), pituitary (prolactinoma, somatotropinoma), and enteropancreatic unit (gastrinoma, insulinoma, pancreatic polypeptide-oma, carcinoid). In addition, neoplasms of the adrenal cortex, neuroendocrine tumors of the bronchopulmonary tree and thymus, lipomas, angiofibromas, collagenomas, and meningiomas develop in affected subjects. The postmortem incidence of MEN1 is 0.25%; in hyperparathyroid subjects 1% to 18%; and in patients with gastrinoma 16% to 38%. Hyperparathyroidism (because of a solitary parathyroid adenoma or tumors within several parathyroid glands or hyperplasia of all four parathyroid glands) is the most common manifestation of MEN1 occurring in more than 90% to 95% of affected patients; it is the most frequent endocrinopathy in children with MEN1, at times developing before 10 years of age. Unlike other forms of hyperparathyroidism, equal numbers of males and females are affected in MEN1. Pituitary tumors secreting prolactin and/or GH often (30%–40% of MEN1 subjects) develop as do gastrin- (Zollinger-Ellison syndrome), insulin, and glucagon-secreting tumors of the pancreatic islets and gastrointestinal system (30%–70% of patients); these neoplasms also occur in children and adolescents with MEN1. Hypercortisolemia in patients with MEN1 may be caused by excessive secretion of adrenocorticotropin by a pituitary adenoma or ectopically by a neoplasm or to a primary adrenal tumor. Thyroid neoplasms occur in 25% of patients with MEN1. Nonendocrine tumors, such as lipomas (34%), intestinal and bronchial carcinoids, and other intestinal neoplasms are reasonably common in subjects with MEN1. Indeed, the dermatological manifestations of MEN1, including angiofibroma (85%), collagenomas (70%) are extremely sensitive and specific indicators of this disease. Patients with MEN1 may also develop a schwannoma or a pheochromocytoma, the latter a tumor most often present in patients with MEN types 2A and 2B. Although most of the tumors that develop in MEN1 are benign but functionally hyperactive, those of pancreatic, intestinal, and foregut origin may be malignant.
| Subtype | Gene | Chromosome | Site of Frequent Tumors | Mutations |
|---|---|---|---|---|
| MEN 1 | MEN1 | 11q13.1 | Parathyroid (90%) | Intron 4 N:5168 G ⇒ A (10%) |
| Enteropancreatic (30%–70%)—gastrinoma, | Codons 83–84 (4%) | |||
| insulinoma, pancreatic polypeptide, | Codons 119 (3%) | |||
| nonfunctional | Codons 209–211 (8%) | |||
| Anterior pituitary (30%–40%)—prolactinoma | Codon 418 (4%) | |||
| GH, ACTH, nonfunctional | Codon 516 (7%) | |||
| Adrenocortical (40%)—diffuse and nodular hyperplasia, adenoma, carcinoma | ||||
| Intestinal—gastric neuroendocrine (10%), | ||||
| Thymic neuroendocrine (2%) | ||||
| Other: facial angiofibroma (85%), collagenoma (70%), | ||||
| lipoma (30%), meningioma (8%) | ||||
| MEN 2 | RET | 10q11.21 | ||
| MEN 2A | MCT (90%) | Codon 634 (Cys ⇒ Arg – 85%) | ||
| Pheochromocytoma (50%) | ||||
| Parathyroid hyperplasia (20%–30%) | ||||
| Cutaneous lichen amyloidosis | ||||
| MEN 2B 3 | MCT (> 90%) | Codon 918 (Met ⇒ Thr – > 95%) | ||
| Pheochromocytoma (40%–50%) Associated: | ||||
| Marfanoid habitus | ||||
| Mucosal neuromas (90%) | ||||
| Medullated corneal nerves | ||||
| Megacolon | ||||
| MEN 2C | MCT (100%) | Codon 618 (> 50%) | ||
| MEN 4 | CDKN1B | 12p13.1 | Parathyroid | |
| Pituitary | ||||
| Testicular/Cervical | ||||
| Renal/Adrenal |
Germline mutations in MEN1 , a 10 exon gene that encodes a 610 aa cytoplasmic and nuclear protein termed menin , have been demonstrated in the majority of patients with familial and sporadic forms of MEN1. Menin regulates cell growth, division, and demise in two sites and through multiple pathways. In the cytoplasm, menin binds AKT1 (protein kinase B—OMIM 164730, chr. 14q32.33) preventing its translocation to the cell membrane where it is ordinarily activated by phosphorylation enabling AKT1 to exert proliferative and antiapoptotic effects; by binding of menin to AKT1, both the cytosolic level of AKT1 and its kinase activity are depressed, thereby suppressing its effects on cell division and survival. Menin enters the nucleus with the guidance of two nuclear localization signals in its carboxyl terminus (aa 479–498, 589–608) where it is involved directly in the regulation of transcription, replication, and survival. By binding directly to JunD, menin blocks JunD-mediated inhibition of transcription of activating protein-1 (and consequently cell division); many of the mutations in MEN1 in patients with MEN1 cluster in exon 4 and interrupt the binding of these two proteins (between menin aa 139–142 and 323–428). By interacting with small mothers against decapentaplegic (SMAD)3 (OMIM 603109, chr. 15q22.3), menin inhibits signaling by transforming growth factor (TGF) β and impairs TGFβ-mediated inhibitory control of cell replication. Interaction of menin with the SMAD 1/5 complex inhibits signaling by bone morphogenetic protein (BMP) 2; menin also inhibits the transcription regulating protein—nuclear factor κB (NF-κB). Further, menin interacts directly with a histone methyltransferase complex and with genes that regulate DNA repair and cell replication and apoptosis, such as tumor suppressor TP53 (p53), CDKN1A , CDKN1B , GADD45A , POLR2A , BBC3 , TP5313 , and FAS. Variants of CDKN1A (OMIM 116899), CDKN2B (600431), and CDKN2C (OMIM 603369), proteins that regulate the cell division cycle, have been associated with nonsyndromic primary hyperparathyroidism.
In subjects with heterozygous germline loss-of-function mutations in MEN1 , unregulated cell growth and tumor formation occur when a second insult leads to loss of MEN1 on the normal allele within susceptible tissues. As with CDC73 (vide supra), the “two hit” hypothesis of tumorigenesis in MEN1 denotes that the patient inherits germline susceptibility to neoplasia superimposed on which is a second insult, leading to loss of heterozygosity for chromosome segment 11q13 and biallelic loss of MEN1 ; the second hit may be deletion of a segment of chromosome 11 that includes 11q13 or a mutation (missense, frameshift) within the wild-type MEN1 allele itself, an observation that also extends to MEN type 1 tumors with somatic mutations in MEN1. Several hundred germline mutations in MEN1 have been identified in patients with MEN1; 25% have been nonsense mutations, 15% missense mutations, 45% frameshift insertions or deletions, with more than 80% leading to the synthesis of an inactive product because of loss of the nuclear localization signal or the ability to bind to JunD or other downstream factor. Especially susceptible germline mutational “hot-spots” in MEN1 are the nucleotide 5168 G ⇒ A transition that results in a novel splice site in intron 4, codons 83 to 84, 118 to 119, 209 to 211, 418, and 516 where collectively mutations have been identified in 37% of patients with MEN1. On either side of many of these sites are segments of repeat DNA sequences of single nucleic acids or of dinucleotides to octanucleotides; this configuration may lead to increased susceptibility to “replication-slippage” because of misalignment of the nucleotide repeat segments during DNA replication permitting deletion or insertion of nucleotides at inappropriate sites. Mutations within the exonic/intronic splice site coding region of MEN1 have been detected in approximately 70% of patients with familial MEN1; likely in the remaining 30% of subjects, there is either deletion of one MEN1 allele or a mutation in one of its noncoding regions. New mutations of MEN1 occur sporadically in 10% of patients with MEN1. Familial autosomal dominant, isolated primary hyperparathyroidism has been variably associated with germ-line mutations (e.g., Val184Glu, Glu255Lys, Gln260Pro) in MEN1 as well. Somatic mutations in MEN1 have also been identified in patients with sporadic, isolated tumors of the parathyroid glands, pancreatic islet cells, anterior pituitary, and adrenal cortex. Clinically apparent disease because of mutations in MEN1 increases with advancing age: at 10 years of age 7% of children with a mutation in MEN1 have a detectable endocrinopathy (primarily hyperparathyroidism); 52% of affected 20-year-old subjects manifest one or more tumors; penetrance increases to 87% by 30 years, to 98% by 40 years, and to 100% by 60 years.
There are several forms of MEN type 2—subtypes 2A, 2B, and familial medullary thyroid carcinoma (2C). MCT is the most common neoplasm encountered in MEN 2A occurring in 90% of patients (OMIM 171400) (see Table 20.5 ) (Thakker et al 2012 ; Arnold & Marx 2008 ). It evolves from antecedent parafollicular or C cell hyperplasia within the thyroid gland. These subjects also develop adrenal medullary pheochromocytomas (50%), parathyroid hyperplasia or adenoma (20%–30%), localized cutaneous lichen amyloidosis (suprascapular pruritic deposits of subepidermal keratin), and partial or complete megacolon. Calcitonin, the secretory product of thyroid C cells, is also produced in very large amounts by MCT; its measurement aids in the diagnosis of MCT and in monitoring the response of this tumor to therapy. In addition to MCT and pheochromocytoma, patients with MEN 2B (OMIM 162300) have a Marfanoid habitus, mucosal ganglioneuromas of the lips and tongue, gastrointestinal ganglioneuromas, medullated corneal nerve fibers, and megacolon, but they do not develop parathyroid disease. Familial isolated MCT is a variant of these disorders. Germline heterozygous gain-of-function mutations in the RET protooncogene underlie the pathogenesis of the type 2 hereditary MENs. RET is a 20-exon gene encoding an 860 aa glycosylated cell membrane tyrosine kinase receptor with extracellular, transmembrane, and intracellular domains that is expressed in tissue of neural crest origin (sympathetic ganglia, adrenal medulla, thyroid parafollicular cells) ( Fig. 20.7 ). The natural ligand of this receptor is glial cell line–derived neurotrophic factor ( GDNF , OMIM 600837). Constitutively activating mutations of one of six cysteine residues in the extracellular domain of RET—codons 609, 611, 618, 620, 630, and particularly codon 634 (Cys ⇒ Arg)—are present in patients with MEN 2A. Loss of but one cysteine residue facilitates receptor homodimerization without ligand binding, thereby activating the RET intracellular tyrosine kinase domain resulting in autophosphorylation of critical tyrosine residues (particularly at codons 1015 and 1062), and subsequent signal transduction. An activating mutation has been identified at codon 918 (Met ⇒ Thr) in more than 95% of patients with MEN 2B; this site lies within the tyrosine kinase domain and this mutation permits signal transduction and neural cell transformation and differentiation in the absence of both ligand binding and receptor homodimerization. The p.Met918Thr MEN IIB-associated RET mutation often arises de novo from sporadic mutations that occur in the germline of an older father, because this mutation confers a “selective advantage” upon the mutated spermatocyte. Other missense mutations of RET associated with MEN 2B have been identified at codons p.Val804, p.Ala883, and p.Ser904. Missense mutations at RET codons 609, 611, 618, 620, and 634 within the extracellular domain and at codons 768, 790, 804, and 891 within the intracellular tyrosine kinase domain have been found in patients with familial MCT. In children, hyperparathyroidism may be an unusual manifestation of the McCune-Albright syndrome (OMIM 174800) because of a postzygotic activating mutation in GNAS . Hyperparathyroidism and nephrogenic diabetes insipidus have been noted in subjects with antenatal Bartter syndrome (OMIM 601678) associated with biallelic variants of SLC12A1 (OMIM 600839), the gene encoding a sodium-potassium-chloride transporter protein.
Ingestion of excessive amounts of vitamin D or calcitriol for therapeutic reasons (treatment of rickets, hypoparathyroidism, or other causes of hypocalcemia), megavitamin intake, manufacturing errors in the preparation of vitamin supplements, or inappropriate fortification of milk are significant causes of vitamin D–mediated hypercalcemia. Topical application of creams containing vitamin D or an analogue (e.g., 22-oxacalcitriol) for treatment of psoriasis might also lead to hypercalcemia—particularly if the urinary excretion of calcium is compromised. Patients with granulomatous diseases (noninfectious: sarcoidosis, berylliosis, eosinophilic granuloma, subcutaneous fat necrosis, inflammatory bowel disease; infectious: tuberculosis, histoplasmosis, coccidioidomycosis, candidiasis, cat-scratch disease) and neoplastic disorders (B-cell lymphoma, Hodgkin disease, dysgerminoma) develop hypercalcemia because of associated monocytic (macrophage and other cells) expression of CYP27B1 and resulting production of calcitriol. Unlike renal tubular cells in which 25-hydroxyvitamin D-1α hydroxylase activity is within mitochondria and the transcription of CYP27B1 is closely regulated by PTH, calcitriol, calcium, and phosphate, in monocytes, this enzyme is microsomal in location and its gene is constitutively expressed and calcitriol synthesis quantitatively determined by the amount of substrate. The monocytic expression of CYP27B1 is very sensitive to stimulation by IFN-γ and its postreceptor signal transducer nitric oxide, as well as by leukotriene C 4 ; it is readily suppressible by glucocorticoids, ketoconazole, and chloroquine. In patients with inactivating mutations of CYP24A1 , calcitriol is unable to be further hydroxylated and excreted.
Patients with acquired immunodeficiency disease may become hypercalcemic by infection with granuloma-forming organisms or by osteoclast-activating cytokines elaborated during the course of this disorder. Elevated serum calcium concentrations have also been recorded in children with congenital hypothyroidism, primary oxalosis, congenital lactase deficiency, trisomy 21, and juvenile idiopathic (rheumatoid) arthritis. In some hypercalcemic children, excessive prostaglandin production may be of pathogenetic significance. Hypercalcemia develops frequently in young subjects with the infantile form of hypophosphatasia, likely a consequence of the dissociation of the rates of low bone formation and normal bone resorption. By the same pathogenetic mechanism, acute immobilization of the rapidly growing child with a femoral fracture or a spinal cord injury results in decreased bone mineral accretion and uncoupling of the interaction of osteoblasts and osteoclasts with an increased rate of bone resorption, leading to hypercalciuria, hypercalcemia, and “acute disuse osteoporosis.” Acute disuse osteoporosis and hypercalcemia can even occur in the immobilized hypoparathyroid or vitamin D–depleted individual. Children and adolescents ingesting a ketogenic diet in an effort to manage refractory epilepsy may develop hypercalciuria and hypercalcemia and experience decline in bone mineralization because of dissociation between the rates of bone accrual and bone mineral reabsorption. Hypercalcemia may occur in children receiving excessive amounts of milk and alkali for treatment of gastritis. It may follow successful bone marrow transplantation in infants with osteopetrosis as functional osteoclasts rapidly reabsorb excess bone mineral.
Oncogenic or malignancy-associated hypercalcemia may be the consequence of synthesis and secretion of osteoclast activating agents, such as PTHrP, (rarely PTH), calcitriol, or cytokines (ILs, tumor necrosis factor [TNF], TGFβ), or it may be caused by direct invasion and destruction of bone by the neoplasm. Although hypercalcemia occurs in less than 1% of children with cancer, it may develop in patients with acute lymphatic and monocytic leukemias, Hodgkin and non-Hodgkin lymphoma, rhabdomyosarcoma, hepatoblastoma, neuroblastoma, and Ewing sarcoma. When the rate of bone resorption exceeds the renal tubular capacity for excretion of calcium, hypercalcemia ensues. Increased intake of calcium and absorbable alkali (milk or calcium containing antacids, such as calcium carbonate) for peptic ulcer disease or as dietary supplements lead to absorptive hypercalcemia, hypercalciuria, and nephrocalcinosis. Parenteral nutrition with excessive calcium or aluminum or too little phosphate can also result in hypercalcemia. Hypophosphatemia of various etiologies leads to hypercalcemia as the body attempts to maintain the calcium × phosphate product over 30. Drugs causing hypercalcemia include: thiazide diuretics increase renal tubular resorption of calcium and decrease plasma volume; vitamin D and analogs increase intestinal absorption of calcium; vitamin A and its retinoic acid analogs stimulate bone resorption; lithium increases the set point for PTH secretion—thereby increasing serum calcium concentrations while lowering urinary calcium excretion and thus mimicking HHC1. In the thyrotoxic subject, hypercalcemia is the result of thyroid hormone–mediated stimulation of osteoclast function and subsequent increase in the rate of bone resorption. Pheochromocytomas and some islet cell tumors may be associated with hypercalcemia, in some instances because of cosecretion of PTHrP. Hypercalcemia in the patient with hypoadrenocorticism is the consequence of continued mobilization of bone calcium, decreased renal glomerular filtration of this cation and its increased renal tubular reabsorption and is reversed by restoration of the eucorticoid state.
During recovery from acute renal failure, serum calcium levels may increase because of mobilization of calcium from ectopic sites, such as muscle in which it had been deposited during the hyperphosphatemic phase of the illness from which it is released by rhabdomyolysis. Hypercalcemia can develop in patients with chronic renal failure caused by a combination of factors including: immobilization, aluminum toxicity, excessive ingestion of calcium-containing antiacids or vitamin D or its analogs, and secondary hyperparathyroidism. After renal transplantation, hypercalcemia is often the result of secondary hyperparathyroidism caused by hypertrophy and hyperplasia of parathyroid chief cells that occurred in response to the PTH stimulatory effects of hyperphosphatemia, hypocalcemia, and decreased synthesis of and response to calcitriol during the period of chronic renal insufficiency. In patients with compromised renal function, mild hypocalcemia and calcitriol deficiency develop when the glomerular filtration rate falls below 80 to 60 mL/min/1.73 m 2 , whereas phosphate retention occurs after the glomerular filtration rate has fallen to 60 to 30 mL/min/1.73 m 2 . The secretion of PTH rises secondarily in these patients in an effort to increase the synthesis of calcitriol, increase renal tubular reabsorption of calcium, raise calcium levels, decrease renal tubular reabsorption of phosphate, and lower phosphate values. Prolonged, uncontrolled secondary hyperparathyroidism can lead to relatively autonomous parathyroid hyperfunction (“tertiary hyperparathyroidism”) and hypercalcemia, primarily in patients with chronic renal failure. Hyperplasia of chief cells is followed by defects in function of the CaSR and loss of effective downregulation of PTH secretion refractory to increased serum concentrations of Ca 2 + . There is an expanded number of monoclonal chief cells in which the expression of CASR and the number of vitamin D nuclear receptors have declined. Secondary and tertiary hyperparathyroidism have occurred in patients with prolonged nutritional vitamin D deficiency rickets and in subjects with X-linked hypophosphatemic rickets receiving large amounts of phosphate. Secondary hyperparathyroidism (in which by definition serum calcium concentrations are normal) also develops in patients with inadequate dietary calcium, impaired intestinal absorption of calcium (lactose intolerance, ingestion of phytates, malabsorption syndromes caused by pancreatic insufficiency or celiac disease), or excessive calcium loss in the urine or soft tissues. Enhanced but transient secretion of PTH may accompany the administration of GH to adolescents with chronic renal failure, likely the result of superimposing upon a high basal rate of PTH secretion further increase in the rate of bone remodeling related to somatotropin and sex hormones. In acutely ill adults, administration of GH has been associated with hypercalcemia as well.
Isolated hypercalciuria in the eucalcemic child may be idiopathic or caused by renal medullary or tubular dysfunction or increased intestinal absorption of calcium, including mutations in genes encoding the vitamin D and CaSRs and soluble adenylyl cyclase, demineralizing disorders, such as juvenile idiopathic arthritis, hyperalimentation, metabolic acidosis, excessive protein ingestion, and diabetes mellitus; hypercalciuria with/without hypercalcemia may be observed in patients with familial forms of hypomagnesemia, several types of Bartter syndrome, and distal renal tubular acidosis.
Evaluation
Evaluation of the child with hypercalcemia requires systematic examination of each of its possible causes (see Fig. 20.5 ). Careful review of the personal and family histories and thorough physical examination precede measurement of serum levels of total and ionized calcium, phosphate, creatinine, intact PTH, and 25-hydroxyvitamin D and of urinary calcium and creatinine excretion; renal ultrasonography is used to detect nephrocalcinosis or renal calculi if hypercalciuria is present. If the serum concentration of PTH is not suppressed in a patient with hypercalcemia, the presence of hyperparathyroidism is to be further considered. If repetitive determinations are consistent with hyperparathyroidism, then its pathogenesis is to be identified. Imaging studies of the neck to be undertaken may include ultrasonography (a parathyroid adenoma is hypoechoic), technetium-99m sestamibi scanning with single-photon-emission CT, dynamic (4D) CT imaging (useful to detect multiple and ectopic parathyroid adenomas), and magnetic resonance imaging (thus avoiding radiation exposure).
When hypercalcemia is mild (total calcium concentration < 12 mg/dL), there may be few, if any, symptoms; thus children/adolescents with mild hypercalcemia are often identified unexpectedly by a screening panel of blood chemistries obtained for another purpose. Hypercalcemia may also be detected during studies for renal calculi, abnormal bone mass, pathological fractures, or during screening of families for associated problems. An elevated serum (total or ionized) calcium concentration in a single specimen may also reflect assay variability and must be verified by repeated determinations in a reliable laboratory. Pseudohypercalcemia is the presence of persistently elevated total calcium concentrations whereas the Ca 2 + is normal and is found in subjects with hyperalbuminemia and other dysproteinemic states. Symptoms attributable to hypercalcemia are independent of its cause and are related to the degree of hypercalcemia and include: intestinal—anorexia, nausea, vomiting, abdominal pain (peptic ulceration, acute pancreatitis), and constipation; urinary—polydipsia, nocturia, and polyuria (calcium acts as an osmotic diuretic whereas hypercalcemia impairs the concentrating function of the distal renal tubule); skeletal—bone pain; nervous system—headache, muscular weakness, impaired ability to concentrate, increased requirement for sleep, altered consciousness (ranging from lethargy and confusion to irritability, delirium, stupor, and coma); on occasion, depression may be the major presenting concern in an adolescent with hypercalcemia. In the toddler and young child, hypercalcemia is manifested by anorexia, constipation, poor weight gain, and impaired linear growth (“failure to thrive”). In a series of 52 children and adolescents with hypercalcemia caused by primary hyperparathyroidism, 80% were symptomatic; the most common symptoms were fatigue/lethargy (35%), depression (14%), headache (35%), nausea (29%), vomiting (23%), and polydipsia (21%). Bone involvement (low bone mass, fractures) was present in 30%. All of the children ( n = 17) with nephrolithiasis in this series were symptomatic. In another series of 44 children and adolescents (26 girls) with primary hyperparathyroidism, mean age at diagnosis was 13 years (range 6–18 years), overall 37 were symptomatic (anorexia, weight loss, malaise, depression) and there were 18 patients with nephrolithiasis. At surgery, 29 patients had a parathyroid adenoma and 11 had hyperplastic parathyroid glands—two of whom had MEN. Several features of hyperparathyroidism in children and adolescents differ from those in adults, including heavier and larger adenomas and higher serum and urinary calcium levels but comparable serum levels of iPTH, phosphate, and alkaline phosphatase.
Evaluation of the hypercalcemic child begins with the historical review during which the family/patient is queried not only about symptoms related to hypercalcemia and its consequences (renal calculi) but also about possibly excessive intake of vitamin D, vitamin A and related compounds (such as retinoic acid for treatment of acne), calcium (perhaps to “prevent” osteoporosis), and alkali, or drugs that affect calcium metabolism (thiazide diuretics may “unmask” hyperparathyroidism by increasing renal tubular resorption of calcium thereby raising borderline calcium concentrations into the hypercalcemic range). The family history is explored for members with known disorders of calcium metabolism (HHC1, hyperparathyroidism, renal calculi) or familial neoplasms (galactorrhea as a sign of a prolactinoma, severe peptic ulcer disease as an indicator of a gastrinoma). Except in extreme instances when hypertension (if normally hydrated) or bradycardia, dehydration, decreased muscular strength, or altered consciousness may be present or in the Marfanoid subject with MEN IIB, physical examination of the hypercalcemic child and adolescent is usually normal. Rarely, is a paratracheal (parathyroid) mass palpable in the hyperparathyroid patient. (Subjects with hypercalcemia caused by subcutaneous fat necrosis have firm to hard, irregular, movable masses scattered about the trunk and extremities. Those with WBS have a typical face whereas those with Jansen metaphyseal chondrodysplasia have characteristic skeletal deformities.)
After confirming the presence of total and ionized hypercalcemia, the urinary excretion of calcium is next measured ( Table 20.6 ). If the PTH concentration is normal or elevated and the calcium excretion is low, it is most probable that the patient has HHC1; this diagnosis can be substantiated by the finding of asymptomatic HHC in one of the parents and further defined by identification of the inactivating mutation in CASR . If the patient is hypercalciuric, other causes of hypercalcemia should be sought. With highly sensitive and specific immunoassays for intact PTH 1-84 in comparison with serum calcium values, separation of patients with hyperparathyroidism from those with other causes of hypercalcemia in whom PTH values are low or subnormal is usually possible. In the absence of secondary hyperparathyroidism (chronic renal insufficiency, malabsorption syndromes, ingestion of thiazide diuretics or lithium), consistently elevated PTH concentrations in the hypercalcemic, hypophosphatemic, hypercalciuric child or adolescent are consistent with primary hyperparathyroidism. Although the diagnosis of primary hyperparathyroidism is usually quite apparent in children and adolescents, there is an occasional patient in whom serum calcium and/or PTH values may not be elevated in a single specimen and in whom repeated measurements of serum and urine calcium and PTH values are necessary before this diagnosis can be established. In 52 children/adolescents with hyperparathyroidism, serum calcium values were normal in 10% and PTH levels in 15%; however, in all subjects, the PTH concentration was inappropriately increased relative to the calcium level. Hypercalcemia, hypophosphatemia, and elevated PTH concentrations were recorded in all 44 children and adolescents (6–18 years of age at diagnosis) with primary hyperparathyroidism in a second series. Osteitis fibrosa cystica, brown tumors (localized nonneoplastic areas of bone resorption composed of osteoclast-like multinuclear giant cells, fibroblast-like spindle shaped cells, and hemorrhagic infiltrates), and subperiosteal and endosteal bone resorption can be detected radiographically in most children with hyperparathyroidism, whereas cortical (distal radial) BMD is likely to be decreased in these subjects. Nonspecific findings in the hypercalcemic subject of diverse etiology include shortening of the QT interval by electrocardiography because the rate of cardiac repolarization is accelerated by increased calcium levels, bradycardia, and first-degree atrioventricular block, and nephrocalcinosis and renal calculi detected by ultrasonography. Serum concentrations of PTHrP should be measured when clinical and laboratory findings are consistent with primary hyperparathyroidism, but PTH values are low and humoral hypercalcemia of malignancy is suspected. When PTH concentrations are low in the hypercalcemic patient, metabolites of vitamin D (calcidiol, calcitriol) should be measured and other causes of hypercalcemia sought.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


