Disorders of HDL Metabolism
Scott M. Lilly
Daniel J. Rader
Observational and epidemiologic studies have strongly implicated a low level of high-density lipoprotein cholesterol (HDL-C) as an independent risk factor for atherosclerotic cardiovascular disease (CVD) (1). A 1-mg/dL decrease in HDL-C has been prospectively associated with a 2% to 3% increase in the incidence of coronary artery disease (CAD). HDL-C has been incorporated into the National Cholesterol Education Program (NCEP) guidelines as a routine screening test for risk of CVD and is recommended for consideration as a target of therapy. There is appreciable interest in identifying the basis of variability in HDL-C levels in the human population, the mechanisms of HDL atheroprotection, and the pharmacologic means of safely increasing HDL-C levels to reduce CVD. The most widely accepted hypothesis regarding the antiatherogenic properties of HDL is that it promotes the uptake of cholesterol from peripheral tissues, including the vascular wall, and returns the cholesterol to the liver for excretion in a process termed “reverse cholesterol transport (RCT).” Additional anti-inflammatory, antioxidant, antithrombotic, and nitric oxide-promoting functions of HDL have been described, each of which could contribute to the protective effects of HDL.
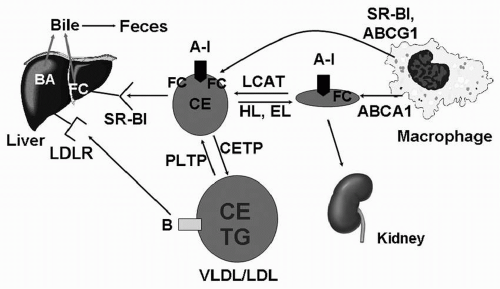 FIGURE 9.1 Structure of the HDL particle. HDL consists of a hydrophobic lipid core of CE and TG surrounded by a shell of PL, free cholesterol, and apolipoproteins such as apoA-I and apoA-II. HDL, high-density lipoprotein; CE, cholesteryl esters; TG, triglycerides; PL, phospholipids; apoA-I, apolipoprotein A-I; apoA-II, apolipoprotein A-II; BA, bile acids; FC, free cholesterol; LDLR, low-density lipoprotein receptor; SR-BI, scavenger receptor type B1; PLTP, phospholipid transfer protein; CETP, cholesteryl ester transfer protein; VLDL, very-low-density lipoproteins; LDL, low-density lipoproteins; LCAT, lecithin-cholesterol acyltransferase; HL, hepatic lipase; EL, endothelial lipase; ABCA1, ATP-binding cassette transporter 1; ABCG1, ATP-binding cassette subfamily G member 1. |
THEORETICAL CONSIDERATIONS
HDL Composition, and Structure, and Heterogeneity
HDL can be differentiated from other lipoproteins (such as low-density lipoproteins [LDL]) by a comparably higher density, smaller size, and the presence of apolipoprotein A-I (apoA-I) (Fig. 9.1). HDL particles are heterogeneous and can be classified based on density, size, or protein composition. The primary structural protein of HDL is apoA-I, which provides a scaffold for the addition of phospholipids (PL) and cholesterol. Triglycerides (TG) constitute a minor fraction of the lipid in HDL. HDL contains a variety of other apolipoproteins, notably apolipoprotein A-II (apoA-II), apolipoprotein A-IV (apoA-IV), apolipoprotein C-I (apoC-I), apolipoprotein C-II (apoC-II), apolipoprotein C-III (apoC-III), and apolipoprotein E (apoE). ApoA-I is present in essentially all HDL particles and represents the majority of HDL protein. Some data suggest that plasma levels of apoA-I may be a better predictor of CVD risk than levels of HDL-C. ApoA-II is the second most abundant HDL apolipoprotein and permits classification of HDL particles into those containing apoA-I but no apoA-II
(LpA-I) and those containing both apoA-I and apoA-II (LpA-I:A-II). LpA-I constitutes about one-third and LpA-I:A-II about two-thirds of HDL particles. The functional differences between these two major classes of HDL particles remain unknown. In addition to the major apolipoproteins, there are a variety of other proteins present in variable amounts on HDL.
(LpA-I) and those containing both apoA-I and apoA-II (LpA-I:A-II). LpA-I constitutes about one-third and LpA-I:A-II about two-thirds of HDL particles. The functional differences between these two major classes of HDL particles remain unknown. In addition to the major apolipoproteins, there are a variety of other proteins present in variable amounts on HDL.
HDL particles can also be classified based on density and size. HDL2 particles have a lower hydrated density (density 1.063 to 1.125 g/mL), a lipid:protein ratio of 60:40, and are larger (10 to 12 nm) than HDL3 particles that have a higher density (1.125 to 1.210 g/mL), a lower lipid:protein ratio (45:55), and are smaller (7 to 10 nm). There is no strict correspondence between classifications based on size or density and those based on apolipoprotein composition. Subsets of HDL-C have not been consistently demonstrated to have different functional effects or to have greater predictive value for CHD compared with HDL-C levels themselves. This underscores the need for better classification systems of HDL, perhaps based on functional rather than structural characteristics.
HDL Metabolism and its Regulation
The metabolism of HDL begins with the secretion of apoA-I from the intestine or liver and the immediate recruitment of cholesterol and PL from these tissues, followed by subsequent addition of lipids and proteins from circulating lipoproteins and peripheral tissues (2). HDL unloads cholesterol and lipid constituents mainly to the liver, and the residual apolipoproteins are eliminated primarily by the kidneys. The distribution of cholesterol and lipid components of HDL and the subsequent elimination of apolipoproteins are key determinants of HDL-C and apoA-I levels and of their variability within the human population.
Biosynthesis of HDL
The preliminary step in forming an HDL particle is the synthesis and secretion of its major apolipoproteins, apoA-I and apoA-II (Fig. 9.2). ApoA-I is produced by the liver and intestine while apoA-II is only produced by the liver. These are under the partial transcriptional control of peroxisome proliferator-activated receptors (PPAR-α). Defects in the biosynthesis of apoA-I or apoA-II confer reductions in HDL-C levels, as discussed in the following text.
ApoA-I is released as a lipid-free protein at risk of rapid renal catabolism and requires PL and cholesterol to form a more stable nascent HDL particle. The immediate recruitment of cellular PL and unesterified (“free”) cholesterol from liver and intestine to apoA-I is mediated by the ATP-binding cassette transporter 1 (ABCA1), expressed on the basolateral membranes of hepatocytes and enterocytes. In an energydependent manner, ABCA1 unidirectionally transfers intracellular cholesterol to lipid-free apoA-I (Fig. 9.2). ABCA1 activity is tightly correlated with HDL-C and apoA-I levels. Subsequent addition of cellular cholesterol to the nascent HDL particle occurs in multiple tissues via the action of ATP-binding cassette transporter 1 (ABCG1) and scavenger receptor class BI (SR-BI), which, unlike ABCA1, prefer larger, lipid-rich HDL2 and HDL3 as acceptors.
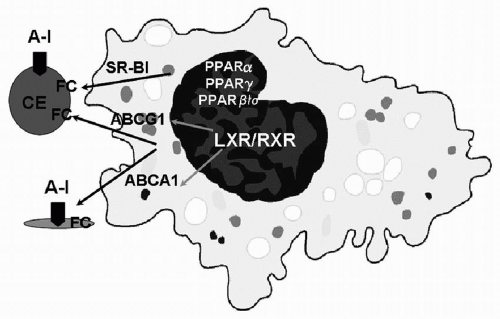 FIGURE 9.2 HDL biosynthesis. Enterocytes and hepatocytes synthesize apoA-I, which is secreted in a lipid-poor form and then immediately recruits additional PL and free cholesterol via the ABCA1 pathway, forming nascent HDL. Nascent HDL acquires more lipid from other peripheral tissues and from lipoproteins, and LCAT generates CE, forming mature HDL. The liver also synthesizes apoA-II, which results in a subclass of HDL containing both apoA-I and apoA-II. HDL, high-density lipoproteins; apoA-I, apolipoprotein A-I; PL, phospholipids; ABCA1, ATP-binding cassette transporter 1; LCAT, lecithin-cholesterol acyltransferase; apoA-II, apolipoprotein A-II; FC, free cholesterol; SR-B1, scavenger receptor class B type 1; CE, cholesteryl esters; ABCG1, ATP-binding cassette subfamily G member 1; PPAR-α, PPAR-γ, PPAR-β/δ, peroxisome proliferator-activated receptors; LXR/RXR, nuclear receptors. |
Acquisition of Lipids and Proteins from TG-rich Lipoproteins
In addition to acquiring lipids from cellular sources, HDL also acquires lipids from triglyceride-rich lipoproteins (TRL) such as chylomicrons and very-low-density lipoproteins (VLDL). Upon the action of lipoprotein lipase (LPL) to hydrolyze TG, TRL shed excess PL and cholesterol, which are then acquired by HDL (Fig. 9.2). LPL is produced by adipose, skeletal muscle, and cardiac myocytes, released to their respective capillary endothelium and tethered by heparan sulfate proteoglycans. LPL is important in regulating the metabolism of HDL and levels of HDL-C indirectly through its action on TRL. The relative contribution of LPL from different tissues to HDL levels and RCT remains a subject of investigation.
Cholesterol Esterification and Maturation of the HDL Particle
The transition of HDL particles from discoidal “nascent” HDL to spherical “mature” HDL involves the esterification of cholesterol to create a hydrophobic core. Lecithin-cholesterol acyltransferase (LCAT) is produced in the liver and found in the circulation associated with HDL particles. LCAT catalyzes the transfer of fatty acids from PL to unesterified cholesterol, forming cholesteryl esters (CE)—an activity enhanced by direct interaction with apoA-I. The CE generated in this fashion comprise the neutral lipid core of mature
spherical HDL3, and further activity of LCAT creates additional CE for the core of HDL3, forming the larger and less dense HDL2 particles (Fig. 9.2). LCAT-mediated esterification of cholesterol is thought to provide a gradient permissive for the transfer of unesterified cholesterol from peripheral tissues to HDL particles and also generates the CE critical for normal HDL maturation.
spherical HDL3, and further activity of LCAT creates additional CE for the core of HDL3, forming the larger and less dense HDL2 particles (Fig. 9.2). LCAT-mediated esterification of cholesterol is thought to provide a gradient permissive for the transfer of unesterified cholesterol from peripheral tissues to HDL particles and also generates the CE critical for normal HDL maturation.
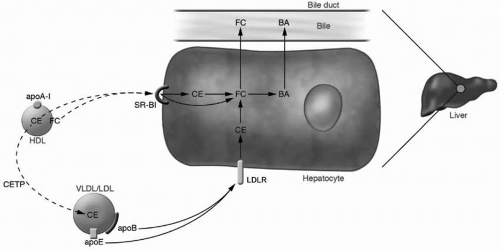 FIGURE 9.3 Pathways of HDL cholesterol uptake by the liver. HDL CE and FC can be directly and selectively taken up by the liver via SR-BI. Alternatively, HDL CE can be transferred to apoB-containing lipoproteins by CETP and then taken up by liver via the LDLR. In the hepatocyte, CE is hydrolyzed to FC, and FC is either excreted directly into the bile or converted to BA and excreted into the bile. HDL, high-density lipoproteins; CE, cholesteryl esters; FC, free cholesterol; SR-BI, scavenger receptor type B1; apoB, apolipoprotein B; CETP, cholesterol ester transfer protein; LDL, low-density lipoproteins; VLDL, very-low-density lipoproteins; LDLR, low-density lipoprotein receptor; BA, bile acids; apoA-I, apolipoprotein A-I. Reproduced from: Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest. 2006;116:3090-3100, with permission. (see color insert.) |
Lipid Transfer between HDL and ApoB-containing Lipoproteins
HDL maturation coincides with the transfer of CE from the core of HDL to circulating TRL. This process is mediated by cholesteryl ester transfer protein (CETP), which exchanges CE from HDL in exchange for TG from TRL. The summation of this process results in the depletion of CE from HDL and enrichment in TG and has important implications for HDL metabolism. CETP activity is one of the most important determinants of HDL-C levels in humans (Fig. 9.3).
Phospholipid transfer protein (PLTP), a protein structurally related to CETP, is produced by the liver and present in human plasma. PLTP mediates the transfer of unsaturated PL from apoB-containing lipoproteins to HDL, thus contributing to the acquisition of PL by HDL (Fig. 9.2). In addition, unsaturated PL are the preferred substrate for LCAT.
Remodeling of HDL by Lipases
Hydrolysis of HDL lipid constituents such as TG and PL occurs extracellularly via the actions of hepatic lipase (HL) and endothelial lipase (EL) (Fig. 9.4). HL is expressed on the endothelial cells of hepatic sinusoids and is upregulated in the insulin-resistant state. HL mediates the hydrolysis of HDL TG, preferably from TG-rich HDL, and in doing so contributes to the conversion of HDL2 to HDL3. EL is produced by endothelial cells in multiple tissues and is upregulated by inflammation and insulin resistance. EL has substantial ability to hydrolyze PL in HDL, also resulting in the generation of smaller denser HDL particles. The activities of both HL and EL result in the release of apoA-I and promotion of its catabolism.
Catabolism of HDL Lipid and Apolipoproteins
HDL CE can be transferred to the liver via the SR-BI (Fig. 9.3). This transfer is “selective,” meaning that the CE is taken up by hepatocytes but not the entire HDL particle, and a relatively CE-depleted HDL particle results. SR-BI also has the ability to mediate selective uptake of free cholesterol from HDL. Hepatic SR-BI is critically important in rodents, which lack CETP, but is of uncertain significance in humans.
The apolipoprotein components of HDL are catabolized at a slower rate than HDL cholesterol and with a variability that exceeds the variability of their production. Together, this implicates apolipoprotein catabolism as a critical determinant of HDL-C levels. Animal studies have revealed that apoA-I is catabolized both in the liver (∼2/3) and in the kidneys (∼1/3). Lipid-poor apoA-I, but not mature HDL, is filtered at the glomerulus, with subsequent internalization and degradation in the renal tubular epithelial cells at a rate directly related to the glomerular filtration rate (Fig. 9.4). The lipidation of
apoA-I and the remodeling of HDL are critical factors that determine the rate of renal catabolism of apoA-I. The mechanisms of hepatic uptake and catabolism of apoA-I are less clear. A subset of large HDL particles contain apoE, which is a ligand for the LDL receptor and may mediate uptake. Other candidates have been identified, including a pathway involving the β-chain of ATP synthase and the G protein-coupled receptor P2Y13; however, they have yet to be proven functionally relevant.
apoA-I and the remodeling of HDL are critical factors that determine the rate of renal catabolism of apoA-I. The mechanisms of hepatic uptake and catabolism of apoA-I are less clear. A subset of large HDL particles contain apoE, which is a ligand for the LDL receptor and may mediate uptake. Other candidates have been identified, including a pathway involving the β-chain of ATP synthase and the G protein-coupled receptor P2Y13; however, they have yet to be proven functionally relevant.
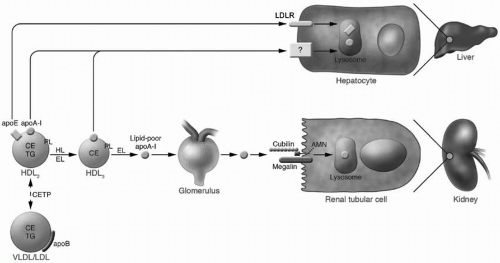 FIGURE 9.4 Pathways of apoA-I catabolism. Lipid-poor apoA-I can be filtered through the renal glomerulus and be degraded by the renal tubular cell via the cubilin-megalin pathway. Lipid-poor apoA-I is generated by the action of lipases such as HL and EL on mature HDL, and the action of these lipases is enhanced by the CETP-mediated transfer of TG into HDL. Mature HDL can also be catabolized by the hepatocyte through binding of HDL-apoE to hepatic receptors or through a poorly characterized mechanism of direct interaction with the hepatocyte. HL, hepatic lipase; EL, endothelial lipase; HDL, high-density lipoproteins; CETP, cholesteryl ester transfer protein; TG, triglycerides; LDLR, low-density lipoprotein receptor; CE, cholesteryl esters; apoA-I, apoB, and apoE, apolipoproteins A-I, B, and E; VLDL, very-low-density lipoproteins; LDL, low-density lipoproteins; PL, phospholipids. Reproduced from: Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest. 2006;116:3090-3100, with permission. |
Antiatherogenic Properties of HDL
The inverse correlation between HDL-C level and CHD is demonstrable at many strata of cardiovascular risk. However, this relationship is not strictly capitulated by a number of HDL-C-raising pharmacotherapies or by the extremes of HDL-C levels that characterize many of the naturally occurring monogenic disorders of HDL metabolism (apoA-IMilano and CETP deficiency, for example). The absolute cholesterol in HDL particles represents a snapshot of the cholesterol pool and fails to capture indices of cholesterol efflux from the vasculature to the liver, as well as the other antiatherogenic functions of HDL. These features of HDL are based on particle composition and enzymatic activity, and methods of evaluating changes in HDL functionality as a result of pharmacotherapy and comparing HDL functionality across the spectrum of CHD risk are being developed (3).
Macrophage Cholesterol Efflux and RCT
Macrophage RCT is thought to be the most important mechanism by which HDL and apoA-I protect against atherosclerosis (2). While macrophages are quantitatively a very small contributor to the pool of HDL cholesterol, cholesterol efflux from macrophages is arguably the most relevant with regard to atherosclerosis. The coexpression and concordant regulation of ABCA1 and ABCG1 by macrophages provides a mechanism for the spatially restricted and sequential addition of lipid to lipid-poor apoA-I and then mature HDL. These have been implicated as the preliminary steps in the reverse transport of cholesterol from macrophages residing in atherosclerotic plaques to the liver. Transcription of ABCA1 and ABCG1 is under the control of the nuclear receptor LXR/RXR (liver X receptor/retinoid X receptor) heterodimer, which is activated by cholesterol derivatives (oxysterols), and likely initiates the homeostatic response to toxic intracellular cholesterol accumulation (Fig. 9.5). ABCA1 transcription is also positively modulated by PPAR-α and PPAR-γ. Posttranslational control of ABCA1 activity occurs via a direct interaction with apoA-I and reciprocally
stabilizes ABCA1 protein, preventing protease-mediated degradation.
stabilizes ABCA1 protein, preventing protease-mediated degradation.
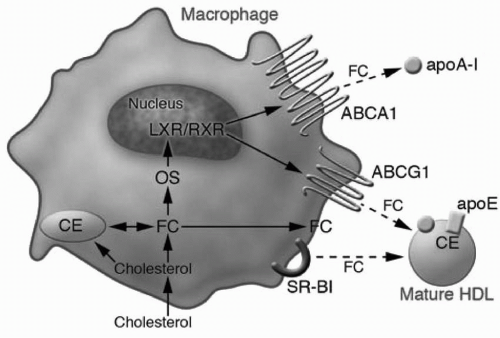 FIGURE 9.5 Pathways of cholesterol efflux from macrophages. Macrophages have several pathways for efflux of cholesterol. They efflux FC to lipid-poor apoA-I via the ABCA1 pathway and to mature HDL via the ABCG1 pathway. Both ABCA1 and ABCG1 are regulated by the nuclear receptor LXR, which is activated by binding of oxysterols, derived from FC. SR-BI also has the capacity to efflux FC to mature HDL. FC, free cholesterol; OS, oxidized sterol; apoA-I, apolipoprotein A-I; ABCA1, ATP-binding cassette transporter 1; HDL, high-density lipoproteins; ABCG1, ATP-binding cassette subfamily G member 1; SR-B1, scavenger receptor class B type 1; CE, cholesteryl ester; LXR/RXR, nuclear receptors LXR/RXR. Reproduced from Rader DJ. Molecular regulation of HDL metabolism and function: implications for novel therapies. J Clin Invest. 2006;116:3090-3100, with permission. |
Completion of the RCT pathway requires transport of cholesterol in HDL to the liver, its uptake by the liver, and subsequent excretion into the bile and feces (Fig. 9.3). Cholesterol in HDL can either be transferred in the form of CE to apoB-containing lipoproteins by CETP (as discussed in the preceding text), or it can be directly and selectively taken up by the liver via SR-BI (as discussed in the preceding text). The relative importance of these two pathways in humans and their relationship to atherosclerosis is still under debate.
Antiatherogenic Effects of HDL beyond RCT
HDL particles exert antiatherogenic effects beyond RCT, including antioxidant, anti-inflammatory, and antithrombotic effects and improvements in endothelial function (4,5). While the contribution of these HDL functions beyond RCT to the prevention and reversal of atherosclerosis remains unclear, strategies for the evaluation of these functions in humans are being developed and are critical to the emergence of directed pharmacotherapies (3). Preliminary events in the formation of an atherosclerotic plaque include endothelial damage with the subsequent infiltration of LDL and recruitment and circulation of monocytes, the latter differentiating into macrophages under the influence of local cytokines. Direct endothelial damage and the release of inflammatory cytokines promotes the expression of adhesion molecules including intracellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule (VCAM-1) on the luminal surface of endothelial cells, which slows circulating monocytes and provides a chemotactic scaffold for the transudation of these cells. HDL are capable of limiting the endothelial expression of ICAM-1 and VCAM-1. When isolated from healthy adults, HDL blunts the cytokine-mediated expression of these endothelial adhesion molecules in human umbilical vein endothelial cells (HUVEC) in vitro, and animal models of direct endothelial injury similarly indicate that HDL infusions blunt the associated expression of ICAM-1 and VCAM-1. The administration of apoA-I mimetic peptides has been demonstrated both in vitro and in vivo to suppress the expression of these cellular adhesion molecules and to blunt atheroma formation. The ability of HDL to abrogate the sequelae of endothelial injury appears at least partially related to its PL composition. The ingestion of a meal high in saturated fat by humans has the proximate effect of attenuating the ability of HDL to suppress ICAM-1 and VCAM-1 expression on HUVEC cells, while a meal high in polyunsaturated fats enhances this function of HDL.
The propensity of monocytes to migrate across the vascular endothelium is also moderated by HDL. The “HDL inflammatory index” is an in vitro index designed to represent the ability of isolated HDL to prevent monocyte chemotaxis, with values >1 representing proinflammatory HDL and values <1 representing anti-inflammatory HDL (6). Although there are presently no prospective reports regarding the relationship between the HDL inflammatory index and subsequent vascular events, there are significant differences in the HDL inflammatory index when populations with and without CHD are compared (1.38 vs. 0.38, respectively). This suggests that proinflammatory (dysfunctional) HDL may play a role in the pathogenesis or maintenance of atherosclerosis in some populations. This notion is supported by the observation that for the subset of patients with CHD and elevated HDL levels (mean 95 mg/dL), the HDL inflammatory index is more representative of the CHD population (1.28) than for its healthy counterpart. Therapeutically, simvastatin reduces the inflammatory index of HDL, offering the possibility that the statins’ effects on HDL underlie some of their imparted cardioprotection. Additional therapeutic strategies are emerging based on animal studies in which the administration of D-4F, an apoA-I mimetic peptide, reduces monocyte chemotaxis and atherosclerotic burden, as does a peptide similar to the apoJ component of HDL.
Oxidized LDL particles are proatherogenic, accumulated by subendothelial macrophages en route to foam cell formation and prompting additional endothelial adhesion molecule expression and monocyte chemotaxis. Serologic markers of oxidized LDL include the proportion of oxidized LDL on apoB-100-containing lipoproteins and antibodies directed against oxidized LDL. These indices bear a significant positive correlation to the presence and extent of angiographically documented CHD and are increased in the setting of acute coronary syndrome. HDL particles can inhibit the oxidation of LDL and abrogate the subsequent inflammatory events preceding atheroma formation and expansion. Implicated enzymes include paraoxonase (PON)-1, glutathione selenoperoxidase, and apoA-I-mediated removal of inciting (oxidation “seeding”) molecules in the subendothelium. Interestingly, HDL can also enhance the oxidation of LDL. These effects can be standardized by assessing the effect of various HDL isolates on the oxidation of LDL PL in vitro. In HDL isolated from humans with CAD, there is a net oxidant effect, while HDL from healthy controls exhibits a net antioxidant effect. Moreover,
simvastatin therapy can temper the oxidant effect of HDL isolates from patients with CHD in vitro, though it does not restore antioxidant activity. These features recapitulate the binary nature of HDL on monocyte chemotaxis and embolden the significance of “proinflammatory” or “dysfunctional” HDL. Similarly, whether HDL serves an antioxidant or oxidant effect on LDL lipid varies with regard to particle composition, local modulators including products of acute inflammation, circulating glucose, and exposure to oxidized LDL itself. Like the anti-inflammatory effects of HDL, antioxidant effects appear to be the default and can be enhanced by the administration of apoA-I mimetic peptides (D-4F) or apoJ, with subsequent reductions in the atherosclerotic burden of predisposed animals (6).
simvastatin therapy can temper the oxidant effect of HDL isolates from patients with CHD in vitro, though it does not restore antioxidant activity. These features recapitulate the binary nature of HDL on monocyte chemotaxis and embolden the significance of “proinflammatory” or “dysfunctional” HDL. Similarly, whether HDL serves an antioxidant or oxidant effect on LDL lipid varies with regard to particle composition, local modulators including products of acute inflammation, circulating glucose, and exposure to oxidized LDL itself. Like the anti-inflammatory effects of HDL, antioxidant effects appear to be the default and can be enhanced by the administration of apoA-I mimetic peptides (D-4F) or apoJ, with subsequent reductions in the atherosclerotic burden of predisposed animals (6).
HDL particles appear also to have favorable effects on the vascular endothelium (5). While normal healthy endothelium produces vasodilatory nitric oxide (NO), which blunts local inflammation, activated endothelial cells release reactive oxygen species, which can precipitate monocyte chemotaxis, platelet aggregation, and atheroma formation. HDL particles promote the formation of NO by vascular endothelium via stimulation of nitric oxide synthase (NOS), an effect contingent upon apoA-I and SR-BI. Parenteral administration of the apoA-I mimetic peptide L-4F also increases NO formation by endothelial cells and blunts oxidized LDL-mediated increases in reactive oxygen species in animal models. Brachial artery vasoreactivity is a commonly utilized surrogate of human endothelial function in vivo, where the arterial dilation is assessed following temporary arterial occlusion (flow-mediated dilation [FMD]). FMD correlates with HDL-C levels in healthy and CHD patients, is comparably blunted in CHD patients, and predictive of future cardiovascular events in high-risk patients. Both HDL infusions and niacin therapy increase FMD in patients with cardiovascular risk factors, though the prognostic significance of this effect has yet to be validated. Additional atheroprotective features of HDL on the endothelium may include increases in endothelial cell proliferation and reductions in apoptosis.
Additional benefits of HDL may manifest in the form of antithrombotic effects (5). In contrast to the effects of LDL, HDL particles inhibit the in vitro aggregation of platelets and the generation of thrombin. Animal models of arterial thrombosis reveal that apoA-IMilano (discussed in the following text) similarly attenuates platelet aggregation and thrombus formation, and human models of venous thromboembolism have identified an inverse relationship between HDL levels and venous thrombosis. The antithrombotic activity of HDL has been linked to its ability to stimulate protein C, thereby inactivating factor Va and arresting the clotting cascade. Whether this is a direct effect or mediated by the ability of HDL to increase the expression of thrombomodulin remains a matter of investigation. Other intermediary candidates include the inhibition of tissue factor, factor X, and the release of plasminogen activator inhibitor. As human models of the antithrombotic effects of HDL emerge, additional candidates are likely to be implicated.
Additional HDL-associated Proteins and their Relevance to Functions beyond RCT
HDL carries a large number of proteins that may influence its function. Many of these have been identified using shotgun proteomics, wherein HDL particles are exposed to protease digestion and the resulting peptide fragments analyzed by mass spectrometry (7). This schema has identified new classes of HDL-associated proteins, including those involved in the acute phase response, the complement cascade, growth factor binding proteins, and proteins involved in hemostasis and thrombosis. Shotgun proteomics has also been utilized to compare the HDL protein composition between healthy controls and those with CHD. α1-Antitrypsin, serum amyloid A (SAA), PON, and fibrinogen are among the acute phase reactants that have been identified in HDL. α1-Antitrypsin is an acute phase reactant and inhibits neutrophil-elaborated elastase, which has been identified in atherosclerotic plaques and implicated in plaque instability. SAA levels also rise appreciably during acute inflammation. These proteins play a role in recruiting neutrophils and monocytes to areas of cellular damage, increasing HDL catabolism, and they have been identified in atherosclerotic plaques where they can tether HDL to extracellular matrix proteins. PON is negative acute phase reactant and believed to moderate the bulk of HDL’s antioxidant capacity. By limiting LDL oxidation, PON blunts a critical preliminary step in formation of the atherosclerotic plaque. HDL-associated PON is reportedly enriched in CHD subjects while serum activity is reduced, the latter being an independent predictor of cardiovascular events. An important remaining question is whether the association of HDL with these acute phase reactants serves a regulatory function, or if HDL is merely a courier for these proteins from the liver to peripheral tissues.
Related posts:
 Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
 Dyslipidemia in Children and Adolescents
Dyslipidemia in Children and Adolescents
 Dietary Treatment of Dyslipidemia
Dietary Treatment of Dyslipidemia
 Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
Use of Noninvasive Methods to Diagnose Cardiovascular Disease in Dyslipidemic Patients
 Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
Dyslipidemia in the Elderly: Special Insights to Inform the Management Strategy
 Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease
Trans Fatty Acids, Dyslipidemia, and Cardiovascular Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


