Introduction
Calcium is vital to many aspects of human biology . For example, calcium is involved in intracellular and intercellular signal transmissions, muscular contraction, coagulation, and bone biology. The body has developed intricate regulatory pathways to ensure a narrow range of concentrations of ionized calcium in the plasma, free calcium in the cytoplasm, calcium incorporation into bone, and calcium absorption by the gastrointestinal (GI) tract. Tightly linked to calcium homeostasis are the regulation of plasma phosphate and the incorporation of phosphate into bone together with calcium . From a biodynamic viewpoint, phosphate-containing compounds and their breakdown are critical in facilitating nearly all energy-dependent cellular processes. Likewise, magnesium (e.g., hypomagnesemia) can impact calcium biology . Magnesium is necessary for normal parathyroid hormone (PTH) release and response to PTH.
After providing an introductory discussion of calcium biology, this chapter deals with three major topics: (1) hypocalcemia, (2) hypercalcemia, and (3) monitoring of bone turnover.
Calcium distribution in the body
The majority of calcium in the body exists in the bone (~98%−99%) . One percent of calcium is in the exchangeable pools. The slowly exchangeable pool includes calcium in atheroma or cartilage (e.g., dystrophic [abnormal] calcification) and calcium in the subcellular pool (e.g., mitochondria and endoplasmic reticulum). The rapidly exchangeable pool is calcium in the plasma, interstitial space, cytosol, and surface calcium in recently deposited bone. Cytoplasmic calcium concentrations are tightly controlled and are approximately 10,000 that of extracellular calcium concentrations. For example, elevations in cytoplasmic calcium concentrations trigger muscular contraction.
Circulating calcium
Calcium exists in the circulation: (1) in a free (ionized) fraction that is biologically active; (2) complexed with anions (e.g., bicarbonate, phosphate, citrate, and lactate); and (3) bound to plasma proteins, predominantly albumin. Approximately 40%–50% of total calcium in the plasma is ionized. The ionized and complexed forms of calcium are filtered by the glomerulus. Protein-bound calcium is not filtered unless there is glomerular disease with loss of pathological amounts of protein.
Because total calcium is significantly influenced by the albumin concentration, if hypocalcemia is identified, albumin should be determined, or, preferably, ionized calcium should be measured to assess to what degree a decline in calcium might reflect hypoalbuminemia and not true hypocalcemia. As an estimate, for each decline in albumin of 1 g/dL, total calcium declines by 0.8 mg/dL, yet ionized calcium will be maintained in the reference interval through the actions of PTH and vitamin D . The differential diagnosis of hypoalbuminemia includes (1) decreased synthesis from liver disease or liver damage or decreased substrate (e.g., malnutrition including malabsorption); (2) chronic inflammatory conditions, infections, or cancer; (3) albumin loss (e.g., protein-losing enteropathy, nephrotic syndrome, burns, and eczematous lesions); (4) dilutional hypoalbuminemia (e.g., posthemorrhage hemodilution or the syndrome of inappropriate antidiuretic hormone); (5) increased catabolism (e.g., thyrotoxicosis, pregnancy, infection, and malignancies); and (6) acute inflammation (albumin is a negative acute phase reactant). Hemoconcentration from dehydration and prolonged tourniquet time can elevate total calcium. Because renal function has a profound effect on calcium metabolism, at the minimum, creatinine must always be measured when assessing calcium and phosphate and certainly when the calcium or phosphate concentrations are outside either of their reference intervals. It can be argued that a blood urea nitrogen measurement and urinalysis should also be included, at a minimum, in a basic renal evaluation as well as calculation of the estimated glomerular filtration rate (eGFR).
Calcium measurements
Clinical laboratories measure ionized calcium using ion-selective electrodes (ISEs), whereas total calcium is measured with dye-binding assays (e.g., arsenazo III, o-cresolphthalein complexone) or by ISE after release of calcium into the free state by the addition of acid. In an ISE, an electrical potential is developed across a membrane that is selectively permeable to a specific ion. For spectrophotometric total calcium measurements, the sample is acidified to release calcium from protein. The o-cresolphthalein complexone-calcium complex then forms at an alkaline pH.
If plasma calcium is measured, the anticoagulant should be heparin (either sodium or lithium). Ethylenediaminetetraacetic acid (EDTA) (e.g., a purple or lavender top tube) binds calcium and prevents its binding to o-cresolphthalein complexone and arsenazo III lowering the measured calcium level. Calcium-EDTA complexes are soluble and can be measured by atomic absorption, although this is rarely performed. Certain forms of gadolinium (used as a radiological contrast agent; specifically gadodiamide and gadoversetamide; gadoteridol, gadopentetate dimeglumine, gadobutrol, and gadoxetate disodium do not reduce measured total calcium) can bind calcium and lower measured calcium without altering the patient’s ionized calcium level. Therefore, some types of gadolinium can cause a “pseudohypocalcemia” .
Ionized calcium can be measured in whole blood (e.g., heparinized blood, either sodium or lithium heparin) using an ISE; however, serum measurements are an alternative. Proper sample handling to avoid stability problems is important. For the measurement of whole blood ionized calcium, the sample should be delivered to the lab on wet ice within 10 min of being drawn. It should be kept refrigerated and measured within 4 h. For serum-ionized calcium, once the blood has coagulated, the sample should be quickly centrifuged (e.g., within 1 h) following the phlebotomy. The sample must be maintained anaerobically until the assay is completed because loss of carbon dioxide from the sample will alkalinize the sample and lower the ionized calcium concentration because pH changes can affect the balance of ionized versus bound calcium. Alkalosis increases the bound fraction, whereas acidosis increases the ionized fraction. Such changes can also occur in vivo; a hyperventilating patient can develop tetany from an acute decline in the ionized calcium concentration. Specimens must also be promptly analyzed or separated from the cells to prevent the change in pH associated with metabolism. The desirable imprecision for calcium based on biological variation is 0.9%, which is much less than that mandated by the Clinical Laboratory Improvement Amendments (acceptable imprecision is ±1 mg/dL) .
It may be necessary to determine urinary calcium (and phosphate or cyclic adenosine monophosphate [cAMP]) excretion in a 24-h urine collection. The 24-h urine excretion of cAMP can be determined as the cAMP urine concentration multiplied by the urine volume. cAMP can also be expressed as ratio of its concentration relative to the creatinine concentration. Standard procedures are used for such measurements.
The upper limit of the reference interval for calcium excretion in men is 300 mg/day and in women it is 250 mg/day. Adults daily excrete about 4 mg of calcium/kg of body weight. To assess the completeness of collection, creatinine should also be measured on a 24-h urine sample. Normal adults excrete 1–2 g of creatinine in the urine daily. The upper limit for the calcium to creatinine ratio in adults is 0.14 mg/mg of creatinine. In children, a calcium to creatinine ratio of ≥0.20 mg/mg is considered to be elevated . In this reference, the correlation of the calcium to creatinine ratio to the 24-h urinary calcium excretion was significant ( P <.001); however, the strength of correlation was modest ( r =0.548). Also of interest, 3–4 year old children had higher calcium excretion (calcium to creatinine ratio: mean 0.16) than children of 5–9 years old (calcium to creatinine ratio: mean 0.10). The tubular reabsorption of calcium (TRCa) can be calculated as 1 minus the fractional excretion of calcium (U=urine; P=plasma):
TRCa=1–[(UCa++/PCa++)×(PCr/UCr)]
Phosphate measurements
Phosphate in serum exists as <SPAN role=presentation tabIndex=0 id=MathJax-Element-2-Frame class=MathJax style="POSITION: relative" data-mathml='H2PO4−’>?2PO−4H2PO4−
H 2 PO 4 −
and <SPAN role=presentation tabIndex=0 id=MathJax-Element-3-Frame class=MathJax style="POSITION: relative" data-mathml='HPO4−2′>HPO−24HPO4−2
HPO 4 − 2
. This inorganic phosphate is measured by these methods: upon reaction of H 3 PO 4 with (NH 4 ) 6 Mo 7 O 24 under acidic conditions, (NH 4 ) 3 [PO 4 (MoO 3 ) 12 ] is formed that absorbs at 340 nm. Under reducing conditions, molybdenum blue is formed that absorbs at 600–700 nm. Xylidyl blue (magnon) or formazan dye can also be used in complexing magnesium. Plasma phosphate concentrations are reported as the plasma phosphorus (the element) concentrations (31 mg/L elemental phosphorus=1 mmol/L phosphate).
The tubular reabsorption of phosphate (TRP) can be calculated as 1 minus the fractional excretion of phosphate:
TRP=1–[(UPhosphate/PPhosphate)×(PCr/UCr)]
Clinical indications to measure calcium, phosphate, and related analytes
Because calcium is involved in many processes, there are many reasons to measure plasma or serum total calcium. Examples of such indications include central nervous system (CNS) problems (e.g., neurotransmission), GI problems (e.g., motility disorders), muscular problems (e.g., muscle contraction), bone or soft tissue disorders (e.g., calcium is a structural component of bone and can be secondarily deposited in injured soft tissue), cardiac arrhythmias (e.g., signal conduction), renal disease (e.g., calcium abnormalities as a cause or consequence), and cases of acute severe illness where acquired (yet transient) hypoparathyroidism is common. Table 10.1 lists examples of these problems. Calcium should be measured when any therapy is used that can alter calcium (e.g., high-dose vitamin D or vitamin D analogs, PTH, bisphosphates, cinacalcet, or calcitonin [CT]).
|
Phosphate should be measured even if the calcium is normal in patients with unexplained weakness, renal disease, malnutrition, refeeding following malnutrition, and when drugs are used that can affect phosphate levels (e.g., vitamin D) . Hypophosphatemia can produce respiratory insufficiency from diaphragmatic failure . Cardiomyopathy from hypophosphatemia has been reported . Dietary phosphate restriction and phosphate binders can decrease plasma phosphate concentrations.
If the calcium and/or phosphate are abnormal, measurements of alkaline phosphatase and PTH are indicated. Alkaline phosphatase is produced by osteoblasts in response to PTH, by the biliary tract, by the intestine, and by the placenta during the third trimester of pregnancy.
In cases of unexplained hypocalcemia, magnesium should also be measured. Chronic hypomagnesemia causes impaired PTH release and impaired PTH responses that can cause hypocalcemia . Causes of chronic hypomagnesemia include dietary deficiency, gastrointesntial disease (e.g., malabsorption, pancreatitis), increased urinary excretion (e.g., diuretics, diabetes or use of drugs such as cyclosporine), alcoholism, certain drugs (e.g., proton-pump inhibitors), and a variety of endocrine and metabolic disorders (e.g., hyper or hypothyroidism, hyper or hypoparathyroidism, primary hyperaldosteronism, and hungry bone syndrome) . Congenital magnesium malabsorption is an extremely rare disorder. Acute pancreatitis or massive transfusion with citrated blood can cause a transient hypomagnesemia.
Measurement of urinary calcium excretion can be clinically important. For example, measurement of urinary calcium excretion is required to establish the diagnosis of familial hypocalciuric hypercalcemia (FHH) . On the other hand, in primary hyperparathyroidism, if a sufficient degree of hypercalcemia is present, hypercalciuria will be present. This presents a risk for nephrocalcinosis and nephrolithiasis.
As noted above, calcium excretion can be expressed as the calcium/creatinine ratio obtained on a spot urine collection, or calcium excretion can be expressed per 24 h when a 24-h urine collection is obtained. It may be necessary to assess vitamin D stores (i.e., 25-hydroxyvitamin D [25-OHD], calcidiol), the most active vitamin D metabolite (i.e., 1,25-dihydroxyvitamin D [1,25-OH2D], calcitriol), or markers of bone turnover (e.g., bone alkaline phosphatase [BAP], deoxypyridinoline, N-telopeptides, C-telopeptides, and osteocalcin). It can be argued that 1,25-OH2D measurements should only be ordered by endocrinologists as these levels require expertise in interpretation.
To properly interprete 25-OHD levels, PTH should be simultaneously measured. For example, if vitamin D intake is deficient, and there is deficient calcium absorption, PTH should be elevated. However, if vitamin D intake is excessive, causing hypercalcemia, PTH should be suppressed. In pathologic conditions of PTH excess, PTH stimulates increased conversion of 25-OHD to 1,25-OH2D. However some patients with hyperparathyroidism do not display frank elevations in 1,25-OH2D presumably due to an ultrashort feedback loop where 1,25-OH2D suppresses its own production. Similarly, reference intervals for 25-OHD and PTH cannot be established without excluding individuals with mild vitamin D deficiency (and subsequent mild secondary hyperparathyroidism due to vitamin D deficiency), which is common in apparently healthy populations.
Calcium and phosphate physiology
To understand the appropriate selection and interpretation of laboratory tests, a systematic review of basic physiology is necessary. Total body calcium is controlled primarily through the regulation of calcium absorption from the GI tract (through 1,25-OH2D). On the other hand, the major mechanism controlling total body phosphate is the regulation of phosphate excretion by the kidney (regulated by PTH, vitamin D, and fibroblast growth factor-23).
Parathyroid glands and parathyroid hormone
The chief cells of the parathyroid glands secrete PTH. The function of mitochondria-rich parathyroid oxyphilic cells (which are derived from chief cells) is unknown . Normally, there are four parathyroid glands in the neck, but the number of glands can vary between three and five, with ectopic glands located even in the mediastinum . The parathyroid gland senses interstitial ionized calcium through its cell membrane calcium-sensing receptor (CaSR; chromosome 3q13.33-q21.1) ( Fig. 10.1 ) . Interstitial ionized calcium reflects plasma-ionized calcium. The CaSR is a 1078-amino acid transmembrane protein with a large extracellular domain that interacts with ionized calcium. There are seven transmembrane domains and a cytoplasmic tail. The CaSR is a member of the G-protein-coupled receptor family .
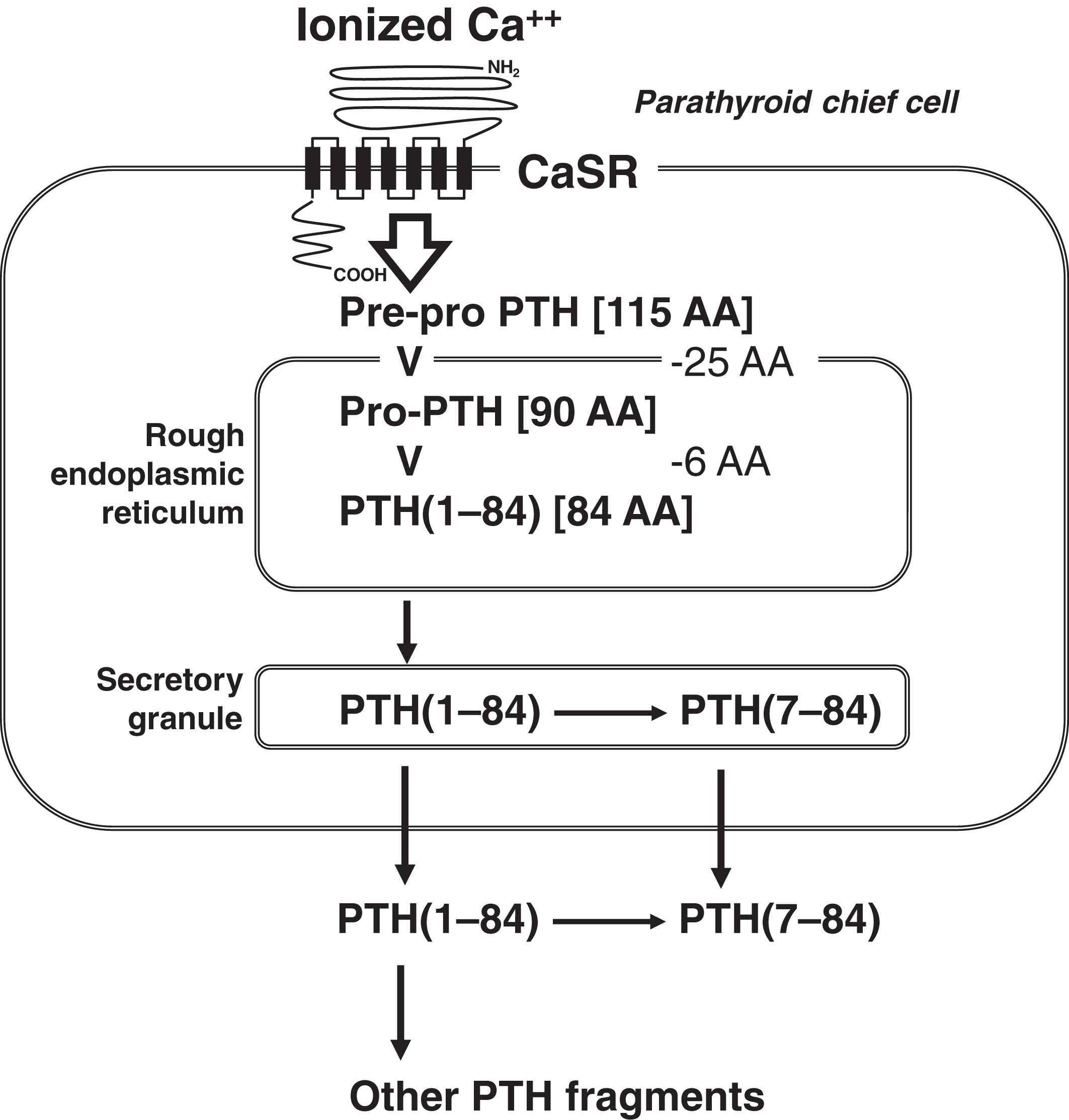
The factors that regulate PTH synthesis and secretion include (1) plasma-ionized calcium; (2) plasma phosphate; (3) 1,25-dihydroxyvitamin D (1,25-OH2D), and (4) fibroblast growth factor-23 (FGF23). Their actions are summarized in Table 10.2 .
| Factor | Effect on PTH release |
|---|---|
| Increased ionized calcium | Decreased PTH |
| Increased phosphate | Increased PTH |
| Increased 1,25-OH2D | Decreased PTH |
| Increased FGF23 | Decreased PTH |
A decline in ionized calcium results in signaling through CaSR with the release of PTH. Elevated phosphate can also release PTH (note: PTH has a phosphaturic effect on the renal tubules). Vitamin D inhibits PTH synthesis. The 5′ regulatory region of the PTH gene has response elements for vitamins D and A. When 1,25-OH2D binds to the vitamin D receptor (VDR; chromosome 12q13.11), this ligand–receptor complex heterodimerizes with the retinoic acid X receptor that serves as a transcription factor binding to the 5′ regulatory region of the PTH gene. Finally, FGF23 serves to lower plasma phosphate and suppresses PTH . This action of FGF23 may relate to the effect of PTH on increasing the formation of 1,25-OH2D, which will raise phosphate through increased GI tract absorption. FGF23 also stimulates 24-hydroxylase (see below) and inhibits the conversion of 25-OHD to 1,25-OH2D. The actions of FGF23 are mediated by its binding to FGFR1 (fibroblast growth factor receptor 1; chromosome 8p11.23) and to, a lesser degree, FGFR4 (fibroblast growth factor receptor 4; chromosome 5q35.2). Klotho (KL; chromosome 13q13.1) is a coreceptor for FGF23 .
PTH is initially synthesized within the cell as a 115-amino acid prepro-PTH molecule ( Fig. 10.1 ). As prepro-PTH enters into the endoplasmic reticulum, the pre- (or leader) 25-amino acid sequence is cleaved. Within the endoplasmic reticulum, pro-PTH (90 amino acids) is cleaved to intact PTH (84 amino acids; PTH[1–84]). Once secreted, PTH(1–84) has a half-life of only ~4 min. Within the parathyroid cell and after secretion, PTH is metabolized to PTH(7–84), N-terminal-truncated PTH fragments of 33 or 36 amino acids, as well as carboxyl (C)-terminal fragments . The concentration of C-terminal fragments is much higher than that of intact or N-terminal-truncated fragments. C-terminal PTH is cleared via the kidney. PTH(7–84), the most prevalent N-terminal-truncated PTH form, appears to have the opposite biologic effect to that of PTH, whereas PTH(1–84) raises calcium, PTH(7–84) lowers calcium and is antiresorptive . PTH(7–84) binds to a C-terminal PTH (C-PTH) receptor . The bioactivity of PTH is contained in its first 34 amino acids.
In the circulation, the ratio of PTH(1–84) to PTH(7–84) varies in health and disease. Usually only 20% of PTH immunoreactivity is PTH(1–84) with the remaining immunoreactivity due to C-terminal fragments . PTH(7–84) is increased in concentration relative to PTH(1–84) in hypercalcemia and chronic renal failure, and is decreased in hypocalcemia. Table 10.3 summarized the actions of PTH and PTH fragments .
| PTH (intact or fragment) | Effect on plasma calcium | Receptor |
|---|---|---|
| 1–84 | Increases | PTH/PTHrP receptor (PTHR1) |
| 1–34 | Increases | PTH/PTHrP receptor (PTHR1) |
| 7–84 | Decreases | C-PTH receptor |
PTH(1–84) normally binds to the PTH receptor-1 (PTHR1), whose gene is located on chromosome 3p21.1–23p21.31. Because PTH-related peptide (PTHrP; see below) also binds to the PTHR1, PTHR1 can also be referred to as the PTH/PTHrP receptor. PTHR1 is a member of the group II subgroup of the G-protein-linked receptor superfamily. The receptor has a large extracellular domain, seven transmembrane domains (e.g., heptahelical), and a cytoplasmic tail similar to the calcium-sensing and thyroid-stimulating hormone receptors. Other examples of this receptor family include receptors for glucagon, glucagon-like peptide I, CT, growth hormone-releasing hormone, secretin, and vasoactive intestinal polypeptide.
PTHrP is produced by some cancers and can cause hypercalcemia through its PTH-like actions. This type of hypercalcemia is termed “humoral hypercalcemia of malignancy.” However, PTHrP is not detected in PTH immunoassays, and separate immunoassays for PTHrP are available from reference laboratories. A second PTH receptor (PTHR2; encoded on chromosome 2q33) binds PTH. Sites of expression of PTHR2 include in the brain, pancreas, testis, and placenta. PTHR2 does not appear to be involved in calcium/phosphate homeostasis .
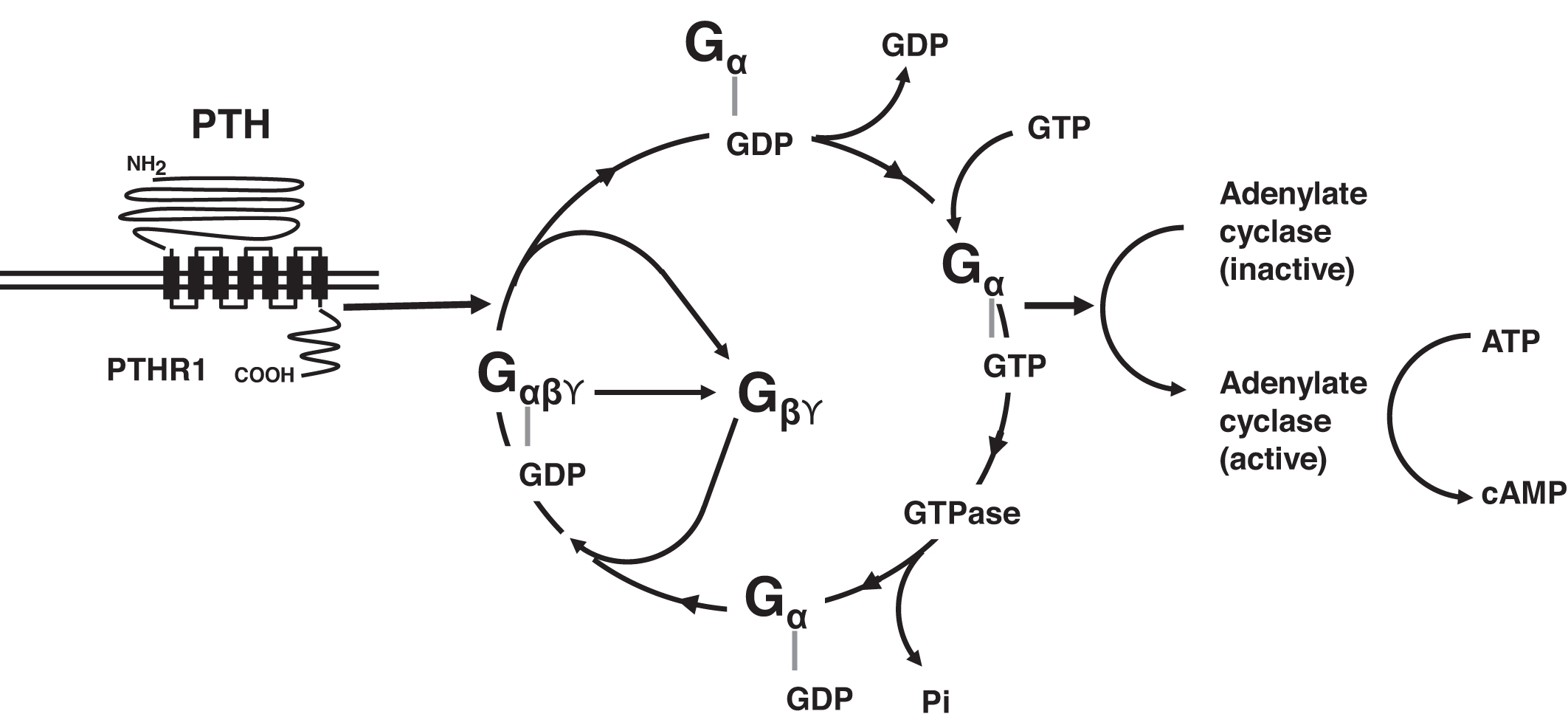
When PTH binds to its receptor, PTHR1 changes conformation and is able to interact with a variety of G-proteins including the guanine-stimulatory (G s ) complex and G q/11 ( Fig. 10.2 ). In the basal state, G s includes G sα , G sβ , and G sϒ . Guanosine diphosphate (GDP) is bound to G sα in this basal state. The interaction of PTHR1 with G s leads to the release of G sβ and G sϒ from G sα , whereas G sα releases GDP and binds guanosine triphosphate (GTP). G sα -GTP next activates adenylate cyclase, converting adenosine triphosphate (ATP) to cAMP. cAMP then carries forward the PTH signal within the target cell. G sα -GTP is self-regulatory: G sα -GTP expresses GTPase activity cleaving a phosphate from GTP yielding GDP. The resulting G sα -GDP recombines with G sβ and G sϒ , and G s in its basal state is reconstituted, and continued stimulation of adenylate cyclase is terminated. PTHR1 activation via PTH also stimulates phospholipase C activity via G q/11 . Other G-proteins include G 12/13 and G i .
Calcium and the renal tubules
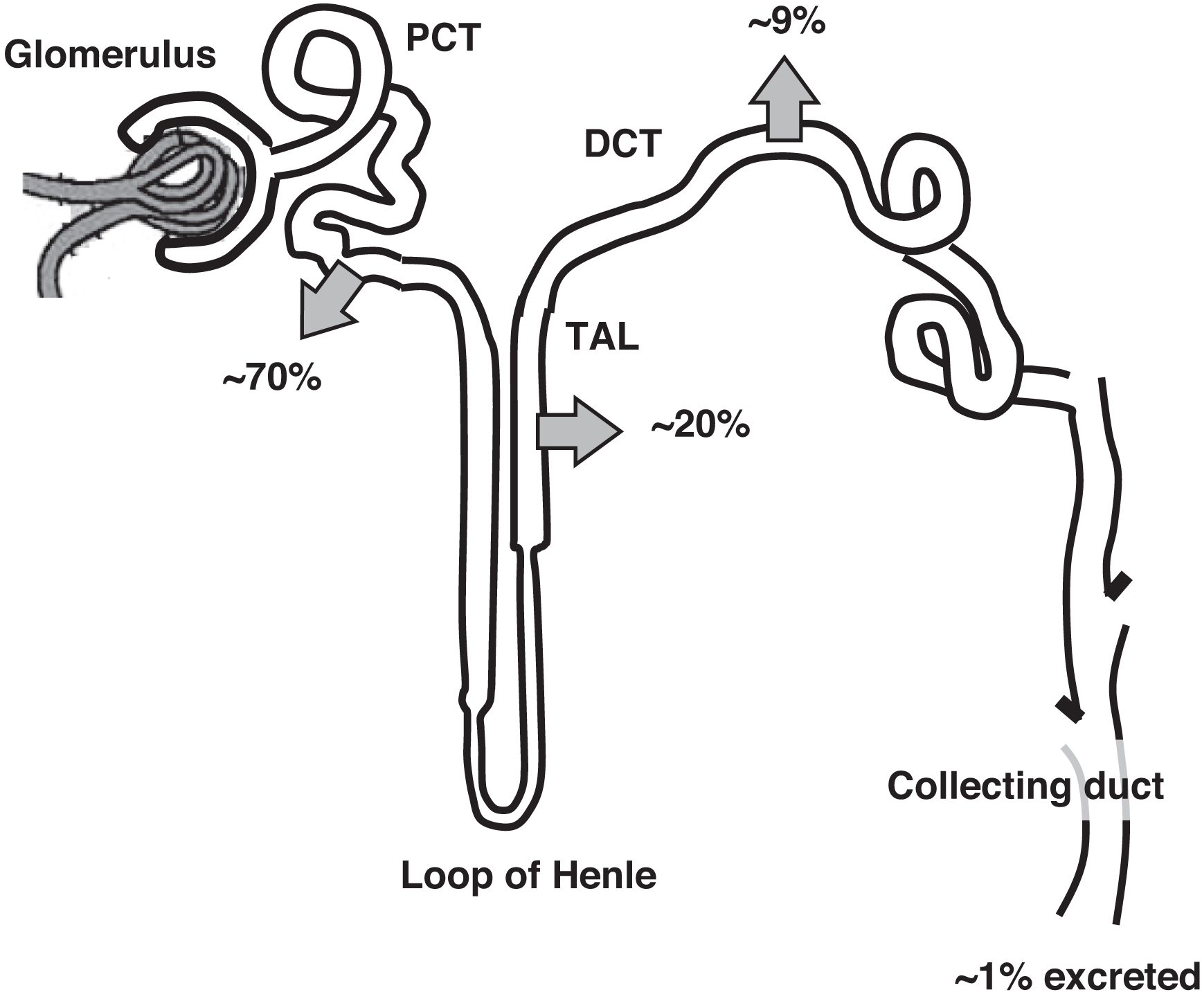
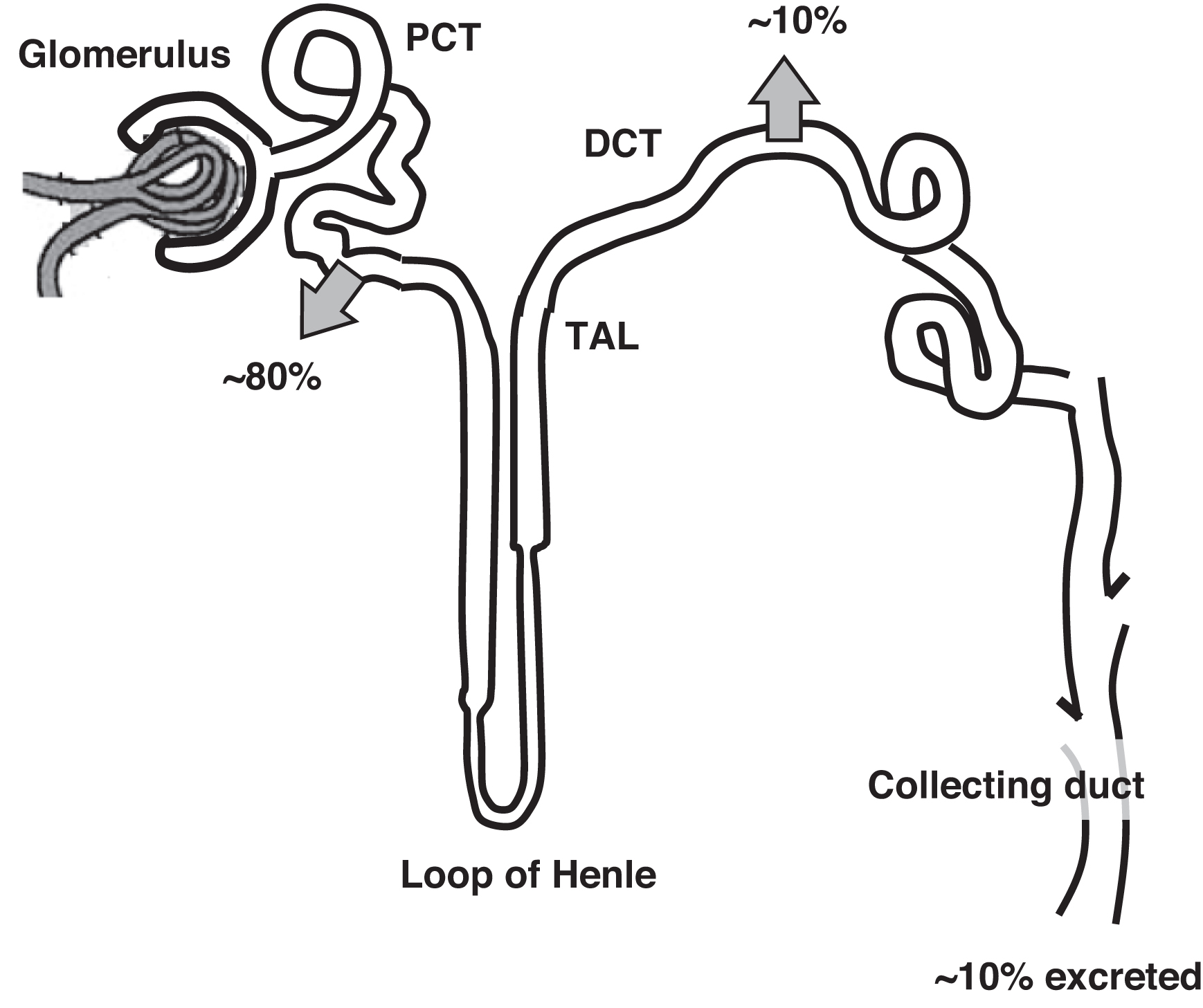
Normally 70% of filtered calcium is reabsorbed in the proximal convoluted tubule (PCT) ( Fig. 10.3 ). The thick ascending loop of Henle (TAL) reabsorbs 20% of filtered calcium with 9% reabsorbed in the distal convoluted tubule (DCT). One percent of filtered calcium is excreted in the urine. In the DCT calcium reabsorption is increased by PTH and 1,25-OH2D. In the PCT and TAL, calcium reabsorption parallels sodium reabsorption. The clinical correlate is that volume depletion will increase renal tubule sodium and calcium reabsorption. In contrast, administering IV saline will encourage calciuria potentially lowering plasma calcium. This is why IV fluids, in part, can help lower plasma calcium concentrations. As well, diluting plasma albumin (with expanded plasma volume from IV fluid infusion) will lower plasma calcium. However, IV fluids would not lower ionized calcium by this mechanism if PTH or vitamin D were in excess.
Loop diuretics, such as furosemide that inhibit NKCC2 (the Na-K-2Cl Cotransporter; also known as SLC12A1 [solute carrier family 12 member 1]; chromosome 15q21.1), lower plasma calcium by increasing calciuria which is secondary to the natriuretic effect of the diuretic. In contrast to loop diuretics, thiazide diuretics that inhibit NCC (Na-Cl Cotransporter; also known as SLC12A3 [solute carrier family 12 member 3; chromosome 16q13]) cause increased calcium reabsorption as DCT sodium wasting elicits increased sodium reabsorption by the PCT and TAL (which increase sodium reabsorption and calcium reabsorption is thus increased).
Cells expressing PTHR1 include renal tubule cells and osteoblasts. In the renal tubular cells, there is increased calcium reabsorption from the renal tubular fluid in the TAL and DCT in response to PTH and 1,25-OH2D. Physiologically, hypercalciuria is to be avoided as hypercalciuria can ultimately cause renal failure from nephrocalcinosis and/or obstruction from nephrolithiasis.
Similar to the GI tract, calcium is reabsorbed from renal tubular fluid through paracellular (between cells) and transcellular (across cells) routes . Paracellular calcium absorption is driven by charge differences through transmembrane tight junctions involving claudins 2 (CLDN2; chromosome Xq22.3), 12 (CLDN12; chromosome 7q21.13), and 15 (CLDN15; 7q22.1) .
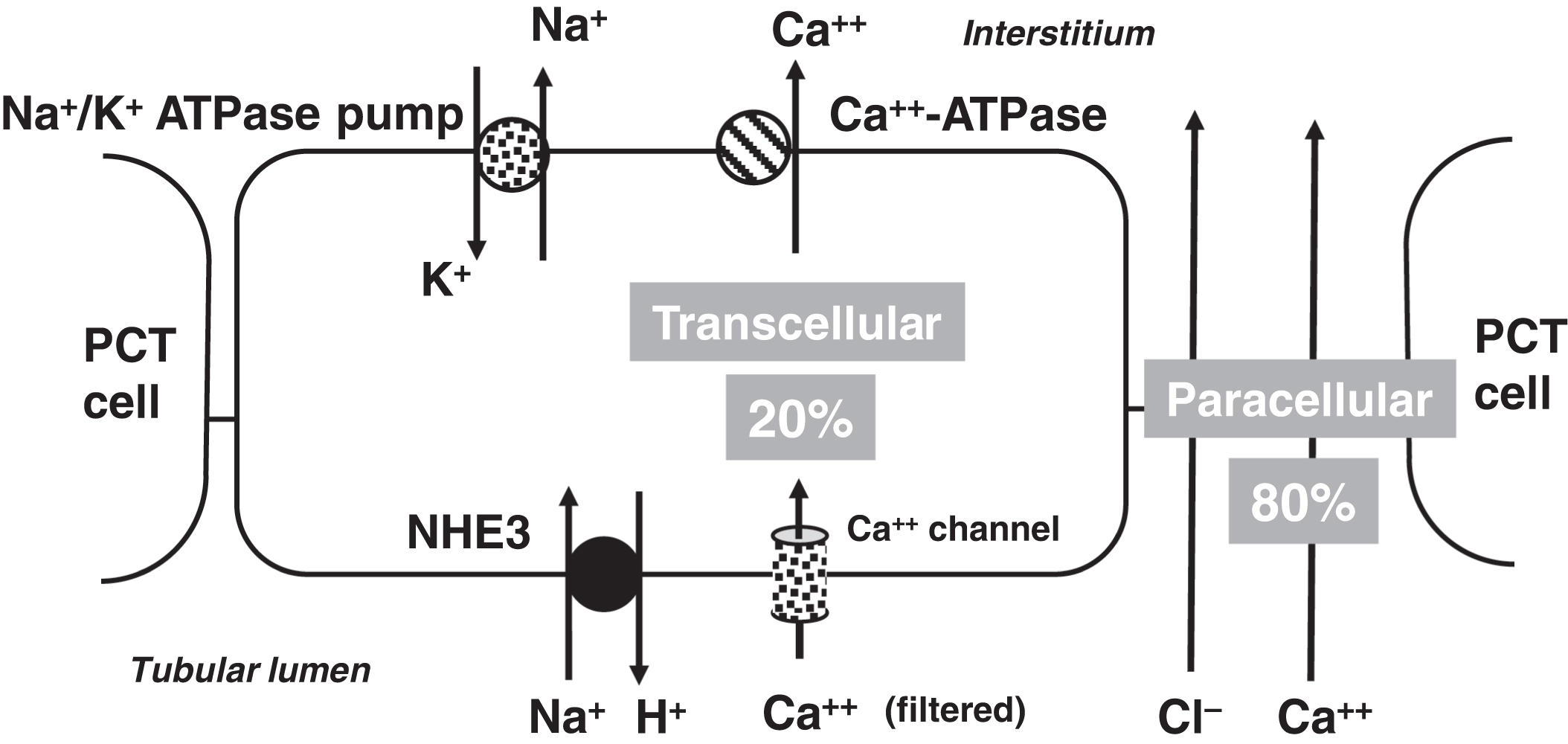
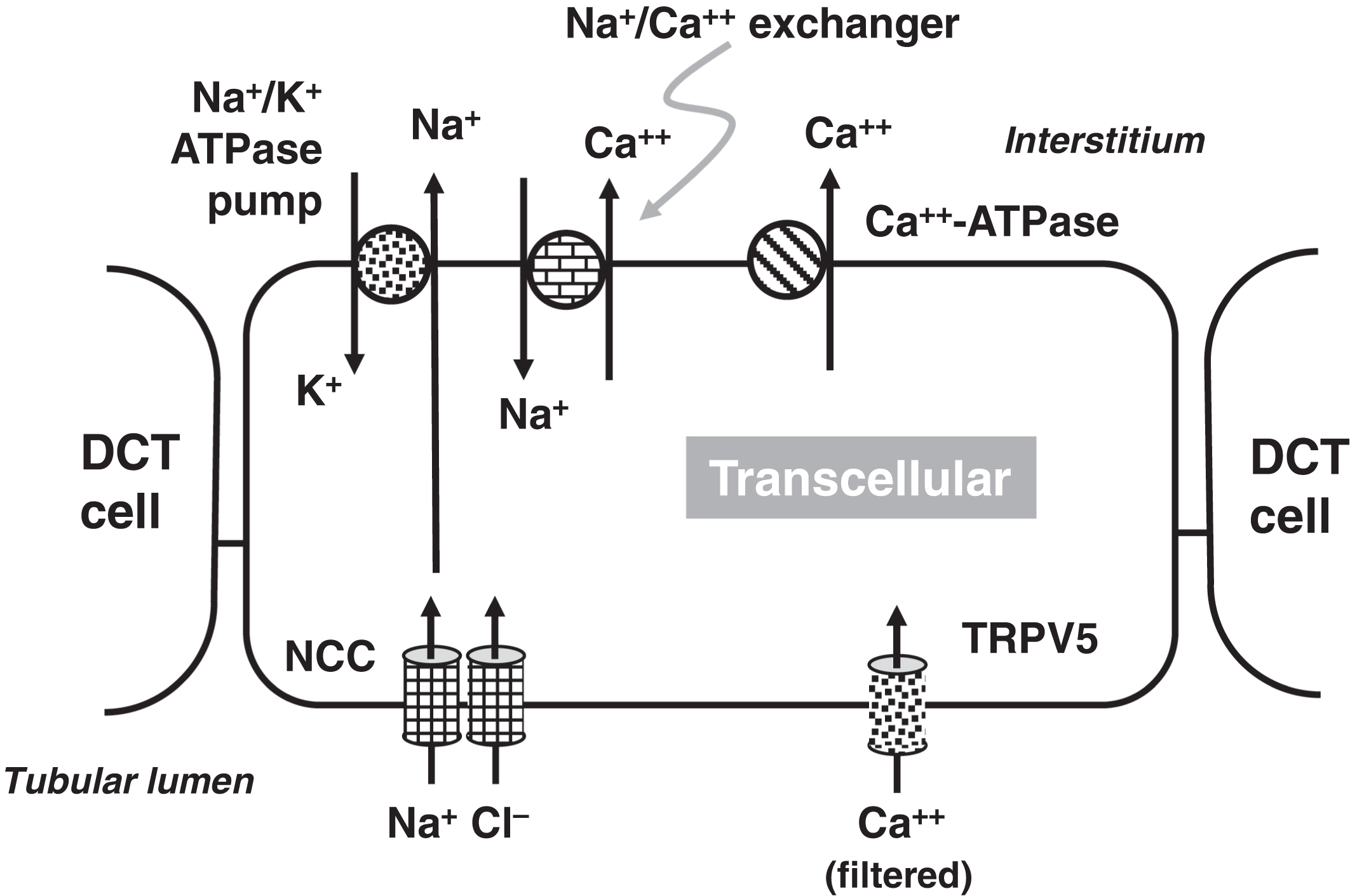
In the PCT, 20% of calcium reabsorption is transcellular utilizing an apical calcium channel and a basal calcium-ATPase pump ( Fig. 10.4 ). Eighty percent of PCT calcium reabsorption is paracellular and is driven by charge. In the TAL, there are both transcellular and paracellular calcium reabsorptions ( Fig. 10.5 ). The transcellular calcium reabsorption of the calcium in the DCT utilizes CaT2 (TRPV5: transient receptor potential cation channel subfamily V member 5; chromosome 7q34) ( Fig. 10.6 ) .
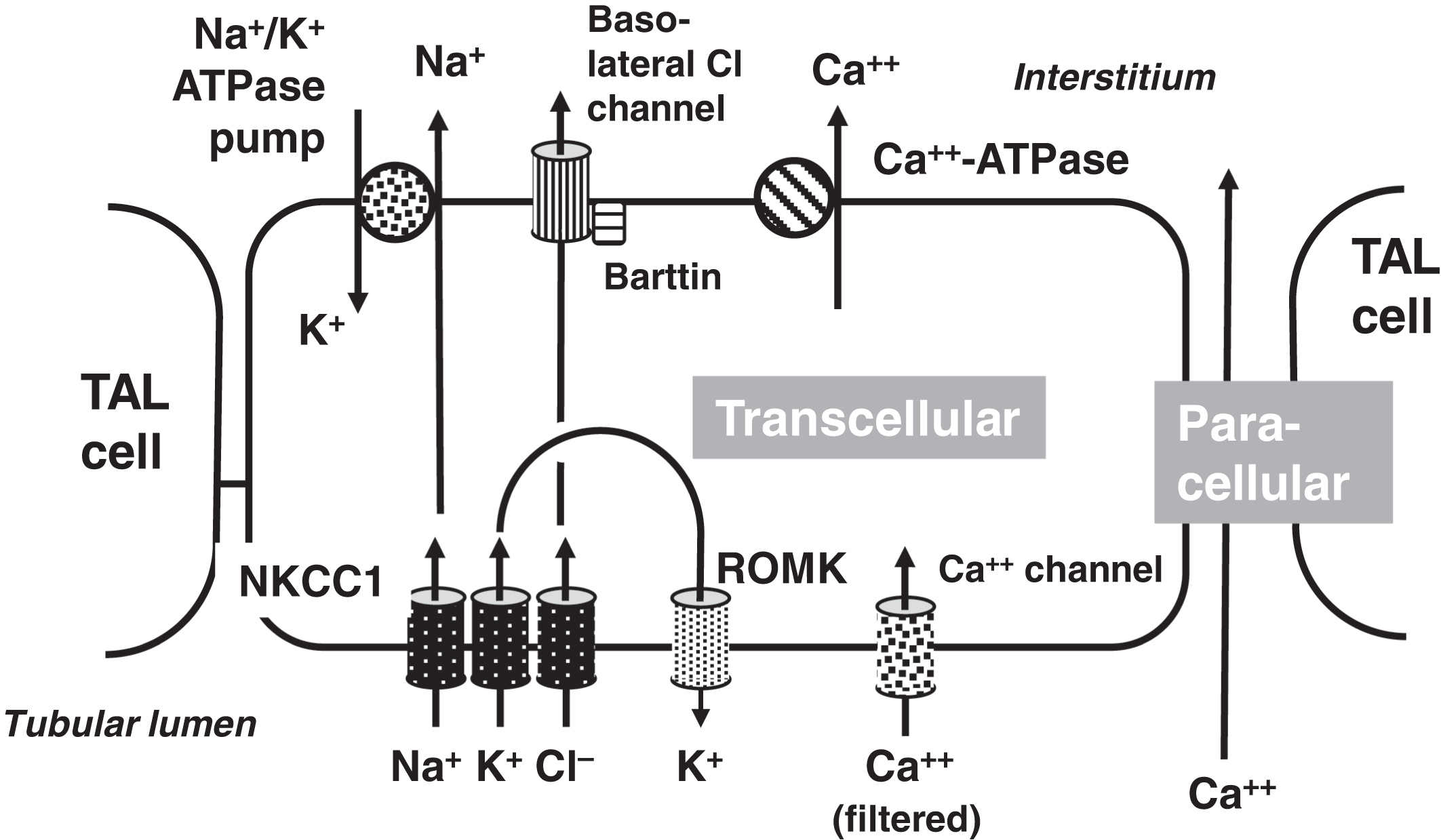
The CaSR is involved in the regulation of urinary calcium reabsorption. The CaSR is expressed in the PCT, TAL, and DCT . The CaSR is most highly expressed in the TAL . When lower plasma calcium concentrations are present, there is reduced stimulation of the CaSR and this leads to increased calcium reabsorption . On the other hand, when plasma calcium concentrations are higher, there is increased stimulation of the CaSR causing calciuria. Therefore, the renal tubular expression of the CaSR provides a mechanism, in part, to regulate plasma calcium independent of the parathyroid glands, C-cells, vitamin D, and FGF23.
Phosphate and the renal tubules
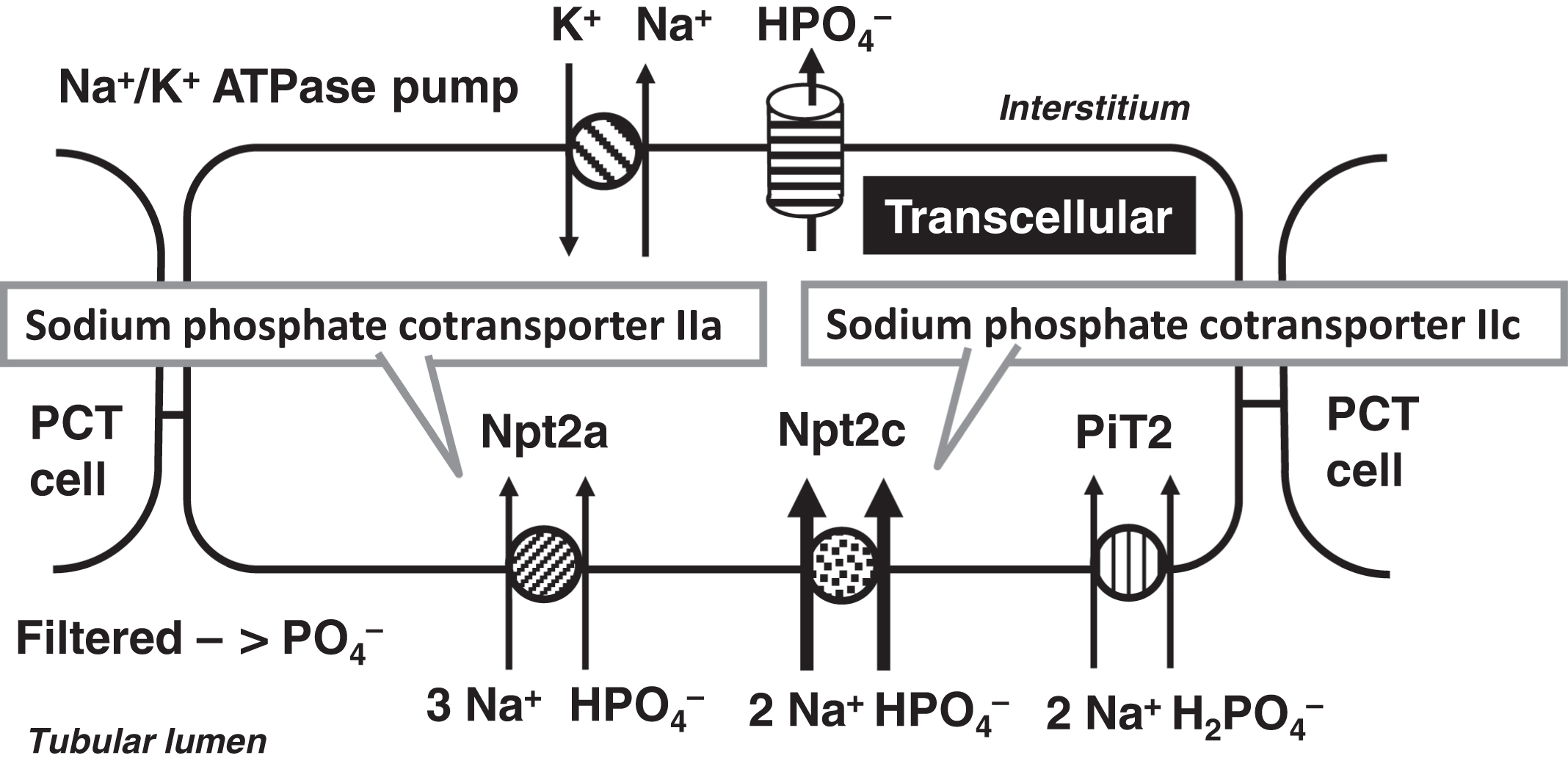
Of the filtered phosphate, ~80% is reabsorbed by the PCT, whereas ~10% is reabsorbed in the DCT and the remaining 10% is excreted ( Fig. 10.7 ). PCT TRP is regulated by PTH, 1,25-OH2D, and FGF23. 1,25-OH2D increases phosphate reabsorption similar to its actions on the intestine. PTH and FGF23 cause increased phosphaturia. These hormones regulate the sodium-dependent cotransporter Npt2c (NaPi-IIa; SLC34A3: sodium-linked channel family 34 member 3; chromosome 9q34.3) and the sodium-phosphate cotransporter Npt2a (NaPi-IIa; SLC34A1: sodium-linked channel family 34 member 1; chromosome 5q35.3) ( Fig. 10.8 ). In humans, Npt2c is more important in phosphate reabsorption than Npt2a . The role of PiT2 (SLC20A2; solute carrier family 20 member 2; chromosome 8p11.21) on phosphate reabsorption is controversial. Several other genes that relate to phosphate homeostasis will be discussed in the section on phosphatemic rickets including PHEX , DMP1 , ENPP1 , and CLCN5 .
Parathyroid hormone and bone
PTH stimulates osteoblasts to build new bone. Stimulated osteoblasts secrete alkaline phosphatase, which is a marker of bone activity. Alkaline phosphatase in growing children is normally two to three times higher than in adults. Adults with healing fractures will have increased alkaline phosphatase levels in the blood. Osteoblasts that become “encased” in ossified bone become osteocytes. Stimulation of osteoblasts will subsequently cause increased osteoclast activity as osteoblast and osteoclast activities are coupled together. Osteoclasts break down the bone. Normal levels of PTH are necessary for healthy bone turnover. However, excessive PTH leads to increased bone breakdown, with a relative abundance of osteoclastic activity compared with osteoblastic activity. Bone is a dynamic biomaterial that in health displays beneficial compromises between lightness and strength, and stiffness and flexibility . Osteopenia, even in the absence of frank osteoporosis, is a major health problem in the elderly that is associated with fractures .
Normal intermittent pulses of PTH stimulate the development of osteoblasts from stromal cells ( Fig. 10.9 ). Stimulated osteoblasts secrete macrophage colony-stimulating factor that stimulates the replication of macrophages into osteoclast precursors.
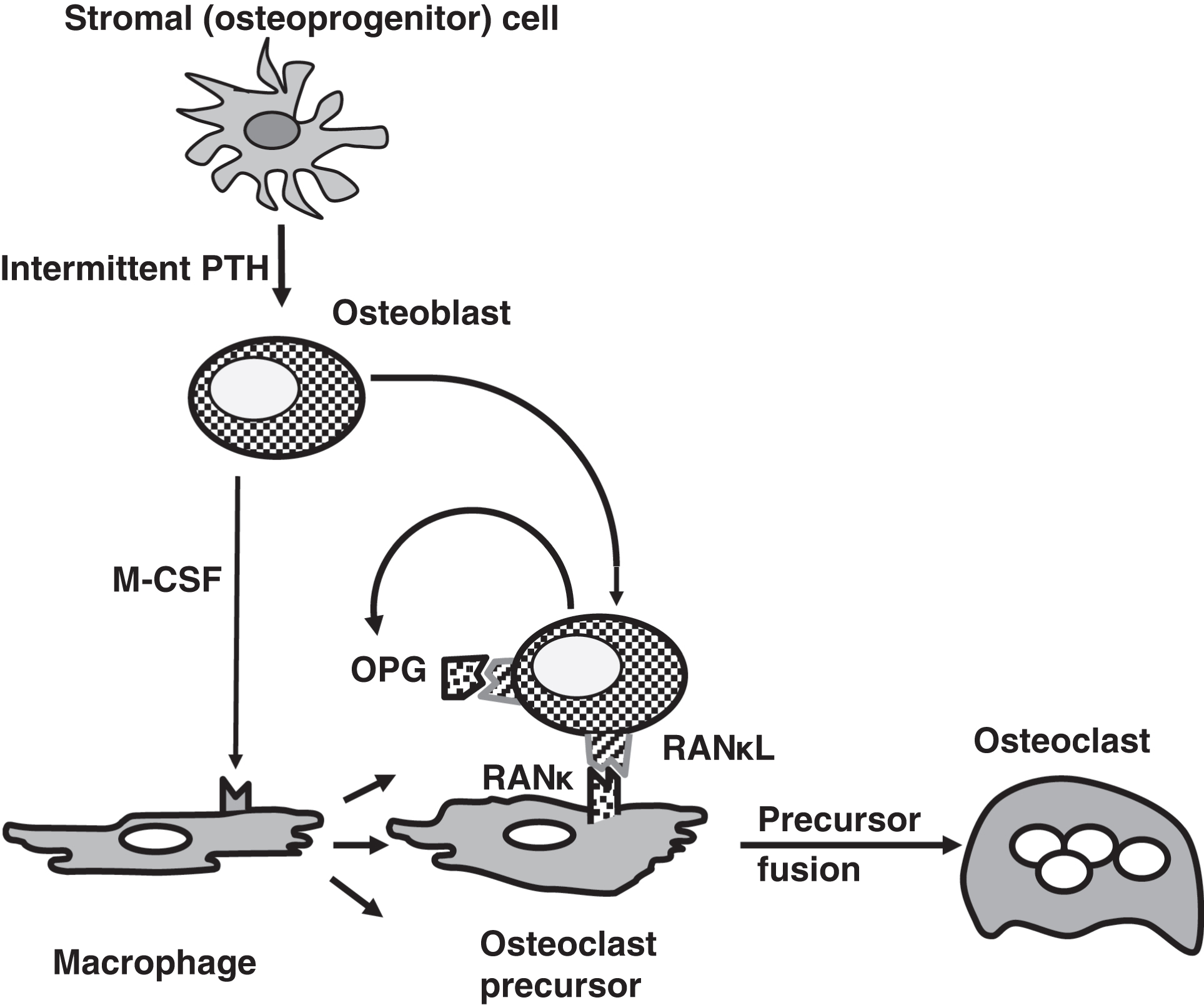
Osteoblasts express the receptor activator of NFκB ligand (RANκL) that binds to receptor activator of NFκB (RANκ) on osteoclast precursors. This stimulates osteoclast precursor differentiation and fusion into osteoclasts. RANκL is also known as osteoclast-differentiating factor. Osteoblasts also secrete osteoprotegerin (OPG) serves as a soluble decoy receptor for RANκL. This binds to RANκL antagonizing the action of osteoblasts in otherwise stimulating osteoclasts. OPG is clinically important as mutations in OPG can cause juvenile osteoporosis. RANκL is clinically important as the monoclonal antibody denosumab can be used to treat osteoporosis.
Calcitonin and bone
The CT gene family includes five members ( Table 10.4 ). The major clinically relevant gene products are CT (323 amino acids) and pro-CT (PCT; 116 amino acids). CT is secreted by the parafollicular clear (C) cells within the interfollicular areas of the thyroid gland. PCT is produced by many tissues; its secretion in inflammatory conditions may be from adipose tissue .
| Gene | Protein product(s) | Chromosome location |
|---|---|---|
| CALCA; calcitonin-related polypeptide alpha | CT, CT gene–related peptide-I (CGRP-I) and katacalcin | 11p15.2 |
| CALCB; calcitonin-related polypeptide beta | CT gene–related peptide-II (CGRP-II) | 11p15.2 |
| CALCP; calcitonin pseudogene | Pseudogene | 11p15.2 |
| AD; adrenomedullin | Adrenomedullin | 11p15.4 |
| IAPP; islet-associated polypeptide | IAPP (also known as amylin) | 12p12.1 |
CT-related polypeptide-I (CGRP-I) is found within the CNS where it may act as a neurotransmitter . Katacalcin lowers calcium . The identification of katacalcin is elusive: one reference identifies katacalcin as amino acids 96 to 106 of PCT that would be within CGRP-I . CGRP-II induces vasodilation of the coronary, cerebral, and systemic blood vessels. Its presence in the CNS also suggests a role as a neurotransmitter or neuromodulator Adrenomedullin is also a vasodilator .
Islet amyloid polypeptide (IAPP; also known as amylin) is produced by pancreatic islet β cells and is cosecreted with insulin. Amylin forms the amyloid deposits seen in and around the islets in about half of the patients with type 2 diabetes who come to autopsy. Amylin injections are available for use therapeutically in the treatment of type 2 diabetes. Pharmacologically, pramlintide, a synthetic analog of amylin, delays gastric emptying and thus can attenuate rises in blood glucose following meals. Pramlintide also suppresses glucagon secretion and centrally regulates food intake.
Concerning CT, osteoclasts are inhibited and plasma calcium is lowered by pharmacological levels of CT. Although CT is a very important hormone in saltwater fish because saltwater contains high levels of calcium, CT in humans has modest physiological importance. For example, if patients receive adequate thyroid hormone replacement following surgical thyroidectomy, bone density will be normal despite CT deficiency. CT deficiency does not cause hypercalcemia.
Serum CT measurements are important tumor markers for the monitoring of residual medullary thyroid carcinoma (MTC) following thyroidectomy . MTC can occur sporadically and predominantly in older individuals. MTC in younger individuals frequently occurs as part of the multiple endocrine neoplasia type 2 (MEN2) , whereas ~100% of persons with MEN2 will develop MTC, 50% will develop pheochromocytoma. Lesser numbers of individuals affected with MEN2 will exhibit hyperparathyroidism. In search of residual MTC following thyroidectomy, provocative testing with calcium or pentagastrin is more sensitive and specific than the measurement of basal CT levels . Familial (autosomal dominant) MTC independent of MEN2 does occur . Familial MTC can be caused by RET proto-oncogene mutations (see below).
As an aside, a drug administered by injection or as a nasal spray, salmon CT (Miacalcin) is Food and Drug Administration (FDA) approved for the treatment of osteoporosis in women who have been menopausal for at least 5 years.
Procalcitonin—a marker of bacterial infection
As noted above, the precursor of CT is PCT. PCT has been proposed as an inflammatory marker indicative of serious bacterial infections . Studies in tissue culture demonstrate that interleukin-1β stimulates PCT production, whereas interferon gamma reduces PCT production . PCT increases within 6–8 h of the onset of fever, plateaus at 12 h, and decreases with antibiotic administration. Thus, PCT is an acute phase reactant; however, its biologic role in inflammation is unclear. It displays a higher sensitivity and specificity for sepsis than C-reactive protein (CRP). Elevations in PCT appear to predict bacterial infection versus a nonbacterial source of infection. PCT also predicts localized infection better than CRP. Elevations in PCT are associated with multiple-organ dysfunction and increased mortality.
The concept for the clinical use of this marker is that lower PCT concentrations suggest a nonbacterial infection not requiring antibiotic therapy improving antibiotic stewardship . However, the value of such testing is controversial . A recent study in more than 1600 patients with lower respiratory tract infections found that PCT testing did not reduce antibiotic use .
Vitamin D physiology
Vitamin D metabolism and actions are critical to calcium and phosphate homeostasis including bone health. The intestine is where total body calcium is regulated and net calcium gain occurs. Vitamin D controls the absorption of dietary calcium and phosphate.
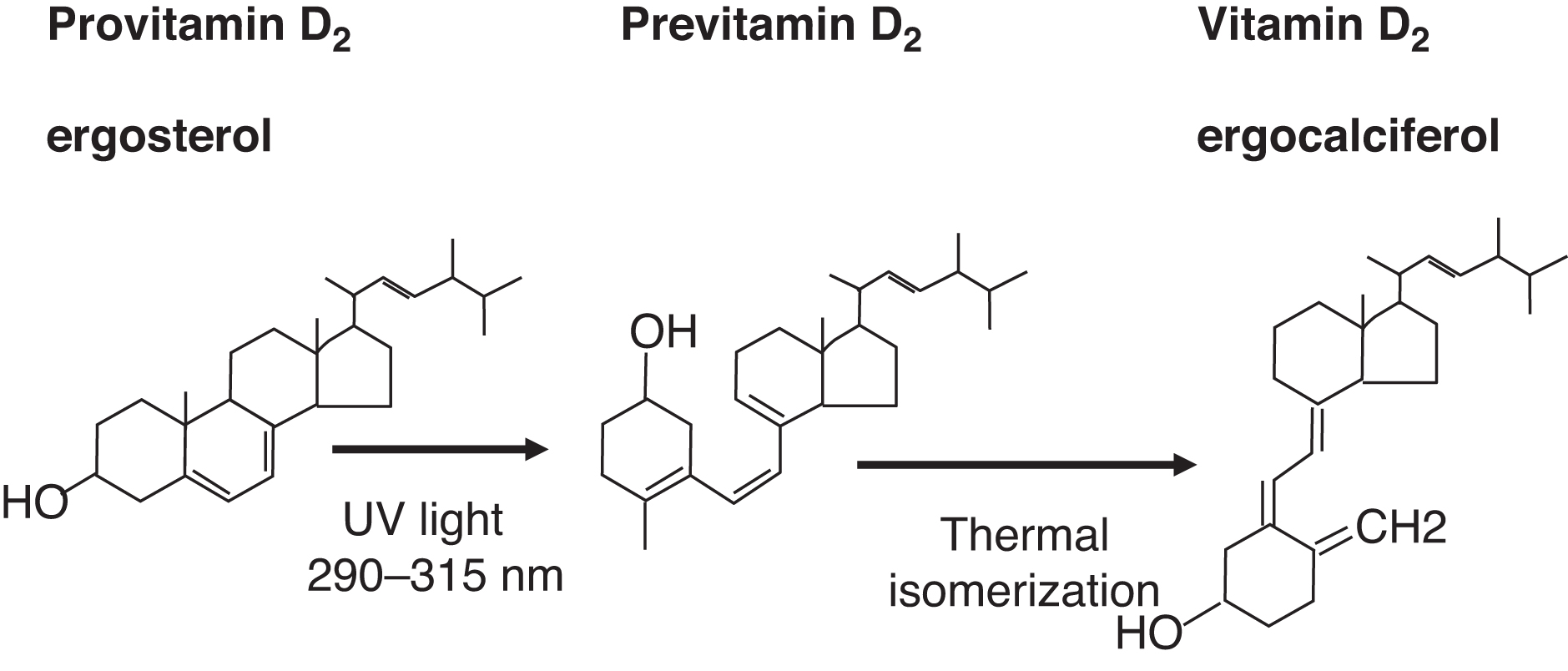
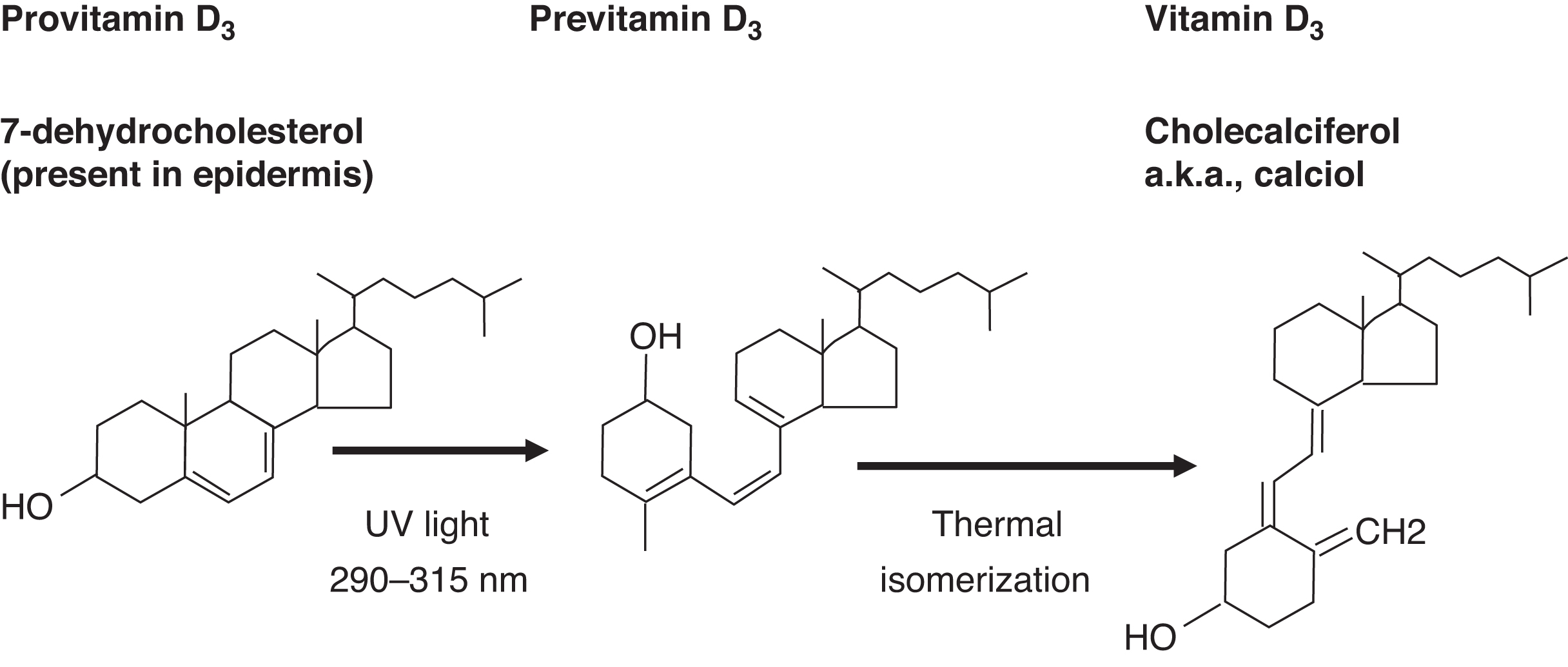
Dietary (e.g., plant) ergosterol is a provitamin D2 compound. Ergosterol is converted by light exposure to previtamin D2 and then isomerized to vitamin D2 (ergocalciferol) ( Fig. 10.10 ). Endogenous 7-dehydrocholesterol in skin is a provitamin D3. Light converts provitamin D3 to previtamin D3, which isomerizes to vitamin D3 (cholecalciferol) ( Fig. 10.11 ).
Because vitamin D is a fat-soluble vitamin, the liver is responsible, in part, for dietary vitamin D absorption. The liver is also the site of conversion of vitamin D to 25-OHD through vitamin D 25-hydroxylase (CYP2R1; cytochrome P450 family 2 subfamily R member 1; chromosome 11p15.2) ( Fig. 10.12 ). Vitamin D stores, measured as 25-OHD, are dependent on ultraviolet light exposure and dietary vitamin D intake.
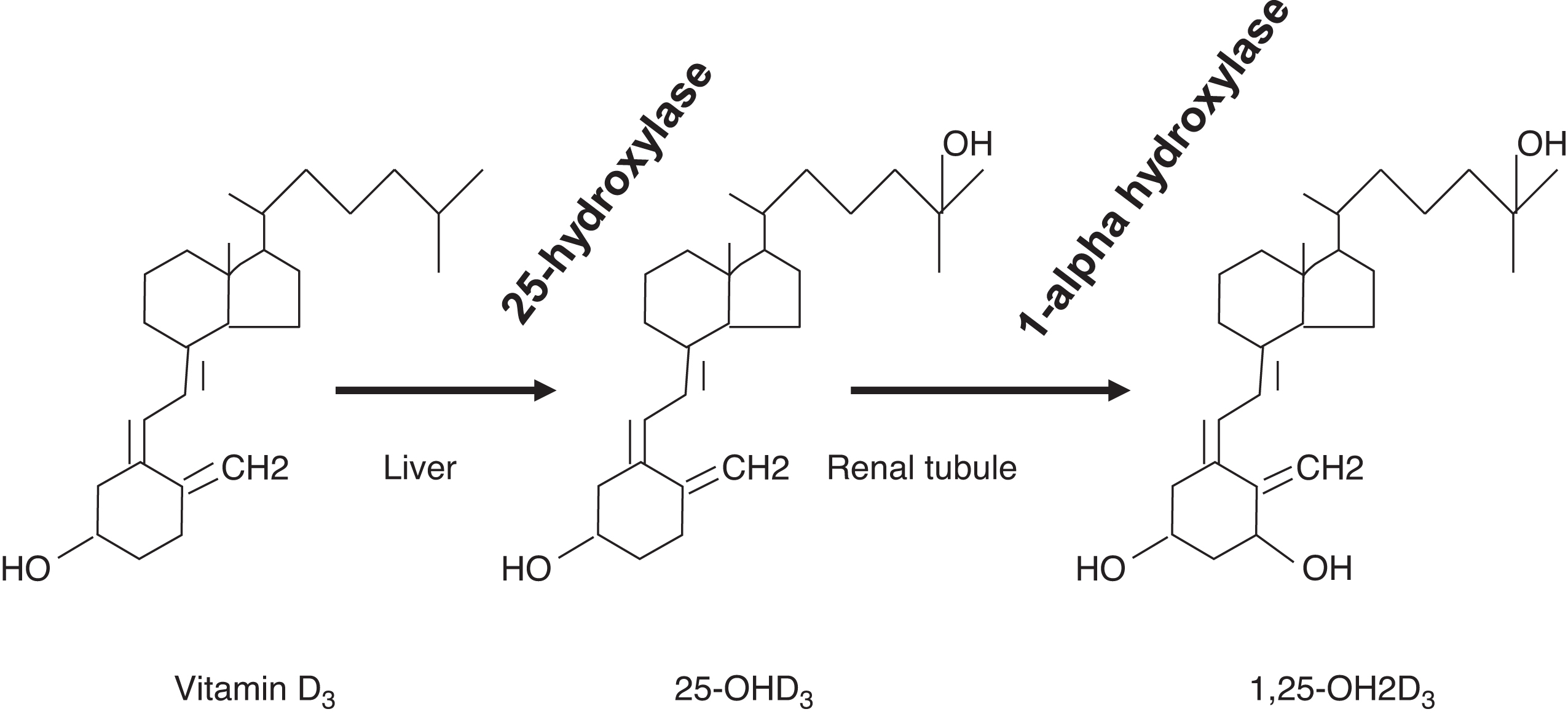
The conversion of 25-OHD to 1,25-OH2D by 25-hydroxyvitamin D, 1α-hydroxylase (CYP27B1; chromosome 12q14.1) is tightly controlled in the renal tubule. The conversion is stimulated by PTH, whereas the conversion is inhibited by elevated phosphate concentrations, elevated concentrations of 1,25-OH2D (i.e., an ultrashort-negative feedback loop), FGF23, elevated calcium, and a high-calcium diet. In addition to increasing the absorption of calcium and phosphate from the gut, 1,25-OH2D is also necessary for normal bone remodeling. Some endocrinologists consider 1,25-OH2D as the active vitamin D “hormone,” whereas 25-OHD is a “prohormone.”
1,25-OH2D is approximately 1000 times more potent than 25-OHD; however, the concentration of 25-OHD is 1000-fold greater than 1,25-OH2D. A typical adult reference interval for 25-OHD is 9–54 ng/mL versus 15–60 pg/mL for 1,25-OH2D. Despite this typical laboratory reference interval for 25-OHD, many experts define vitamin D deficiency as a 25-OHD of <20 ng/mL, relative insufficiency as 20–29 ng/mL, and vitamin D sufficiency as 30 ng/mL or more . However, what constitutes a “healthy” range for 25-OHD is very controversial .
Vitamin D toxicity is only observed when levels of 25-OHD exceed ~100 ng/mL. From a pharmacologic perspective, extremely high-dose oral vitamin D (e.g., 4000 units/day to 10,000 units/day) reduces bone mineral density (BMD) .
Although there has been enthusiasm for a role for vitamin D in immunity , as of 2011, the Institute of Medicine’s (IOM) literature review did not support a role for vitamin D in immunity . The IOM concluded that the only proven physiological role for vitamin D is in bone health. Needless to say, this is frequently challenged in the literature .
25-OHD and 1,25-OH2D metabolism
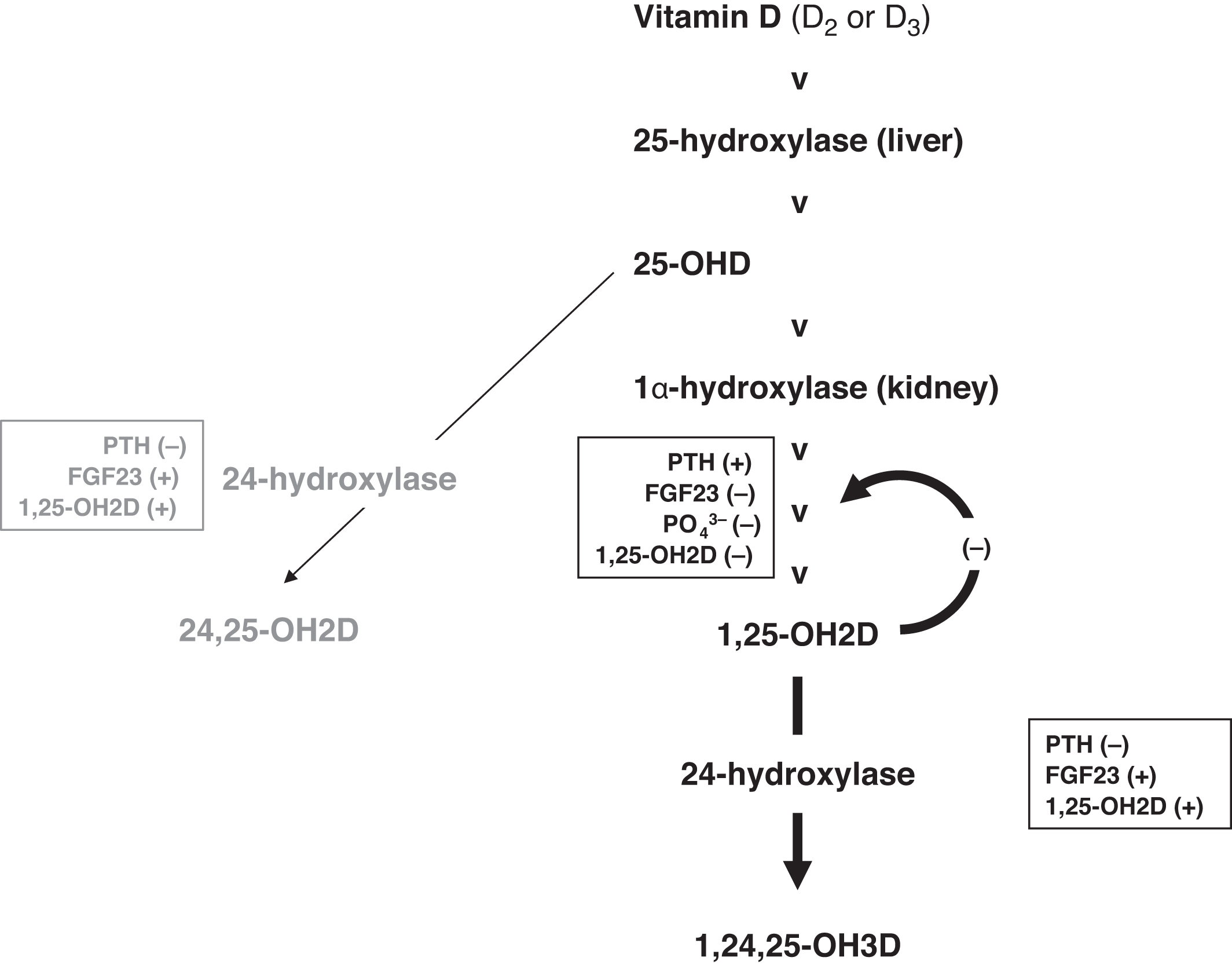
An emerging area of interest is the measurement of the metabolites of 25-OHD and 1,25-OH2D ( Fig. 10.13 ). 25-OHD can be converted to 24,25-dihydroxyvitamin D (24,25-OH2D) by the action of 24-hydroxylase (CYP24A1; cytochrome P450 family 24 subfamily A member 1; chromosome 20q13.2). 24,25-OH2D does not have substantial vitamin D activity. However, a recent study suggests that 24,25-OH2D assists in fracture healing involving the transmembrane receptor FAM57B2 (family with sequence similarity 57, member B) .
PO 4 3 −
) and 1,25-OH2D.
24-hydroxyase also acts on 1,25-OH2D converting 1,25-OH2D to 1,24,25-trihydroxyvitamin D (1,24,25,-OH3D), which is inactive similar to 24,25-OH2D (e.g., ~absent binding to the VDR). This is referred to as the C24 oxidation pathway. Therefore we have two pathways that oppose one another: 25-OHD, 1α-hydroxylase increases 1,25-OH2D; whereas, 24-hydroxylase reduces 1,25-OH2D concentrations.
While increasing the activity of 25-OHD, 1α-hydroxylase, PTH also suppresses 24-hydroxylase . Similar are the actions of phosphate deficiency in raising 1,25-OH2D concentrations and reducing 24-hydroxylase activity. The actions of FGF23 and 1,25-OH2D are opposite to those of PTH (e.g., reduced conversion of 25-OHD to 1,25-OH2D and increased 24-hydroxylase activity inactivating 25-OHD [to 24,25-OH2D] and 1,25-OHD [to 1,24,25-OH3D]). Important regulators are the ultrashort-negative feedback loop with 1,25-OH2D suppressing its own form formation and the conversion of 1,25-OH2D to 1,24,25-OH3D through 24-OHylase.
There is an evidence for a 23 lactone pathway where 1,25-OH2D is converted to 1,23,25-trihydroxyvitamin D (1,23,25-OH3D; also known as 1α,25-(OH)2D-26,23-lactone) through 24-hydroxylase . However, this pathway may not be physiologically important.
Because of the tight regulation of 1,25-OH2D concentrations, because of various compensations, 1,25-OH2D is usually within the reference interval even in deficiency states . One can then conclude that there are few clinical situations where 1,25-OH2D should be measured. Possibly the best use of 1,25-OH2D measurements is in the evaluation of persons with suspected vitamin D–dependent rickets (VDDR) (see below).
Calcium and phosphate absorption from the gut
Transcellular dietary calcium absorption involves three steps ( Fig. 10.14 ). First, calcium enters the intestinal brush border cells through a calcium transporter (CaT1; also known as TRPV6: transient receptor potential cation channel subfamily V member 6; chromosome 7q34). To a lesser extent, CaT2 (SLC7A2; solute carrier family 7 member 2; chromosome 8p22) is responsible for transcellular calcium absorption . Cav1.3 (CACNA1D; calcium voltage-gated channel subunit α1 D; chromosome 3p21.1), an apical L-type calcium channel, may also provide for calcium absorption by the intestine
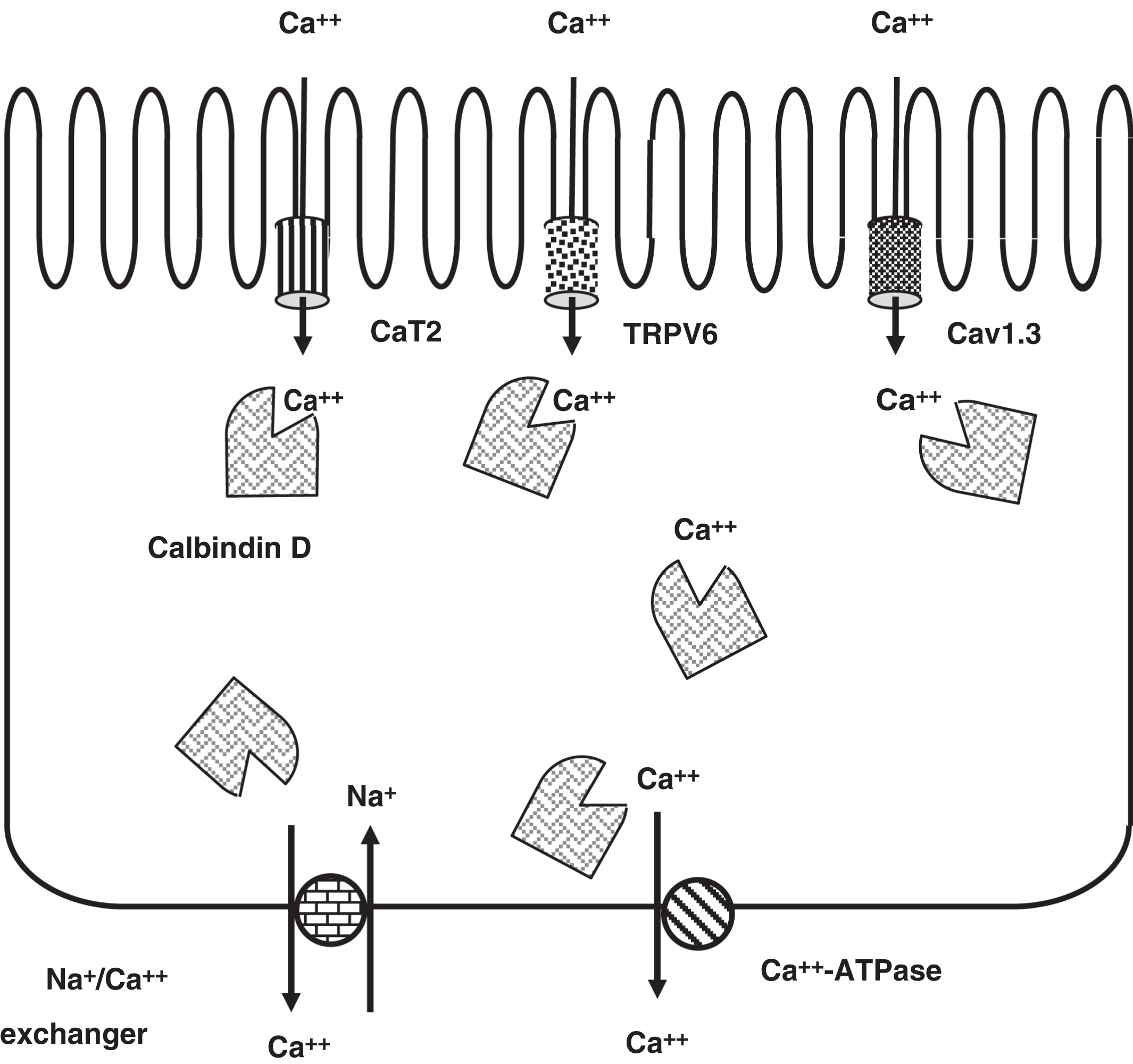
Next, the 9-kDa cytosolic calcium-binding protein calbindin D (CALB1; calbindin 1; chromosome 8q21.3) mediates calcium diffusion within the cell. This allows the cytoplasmic calcium concentration to remain physiologically low. Finally, calcium is pumped out of the cell by a basolateral membrane calcium ATPase (ATP2B1; ATPase plasma membrane Ca 2+ transporting 1; chromosome 12q21.33) and a Na + /Ca 2+ exchanger (SLC8A1: solute carrier family 8 member A1; chromosome 2p22.1). The first two steps are rate limiting in the process of calcium absorption; TRPV6 (CaT1) synthesis is ~90% dependent on 1,25-OH2D, whereas calbindin D synthesis is completely dependent on 1,25-OH2D.
Usually, about 70% of dietary phosphate is absorbed. This is dependent upon the actions of 1,25-OH2D on the gut. There is a sodium-phosphate transporter expressed in the gut apical membrane: NaPi2b (SLC34A2; see discussion of hypophosphatemic rickets below).
Integrating parathyroid hormone and vitamin D actions
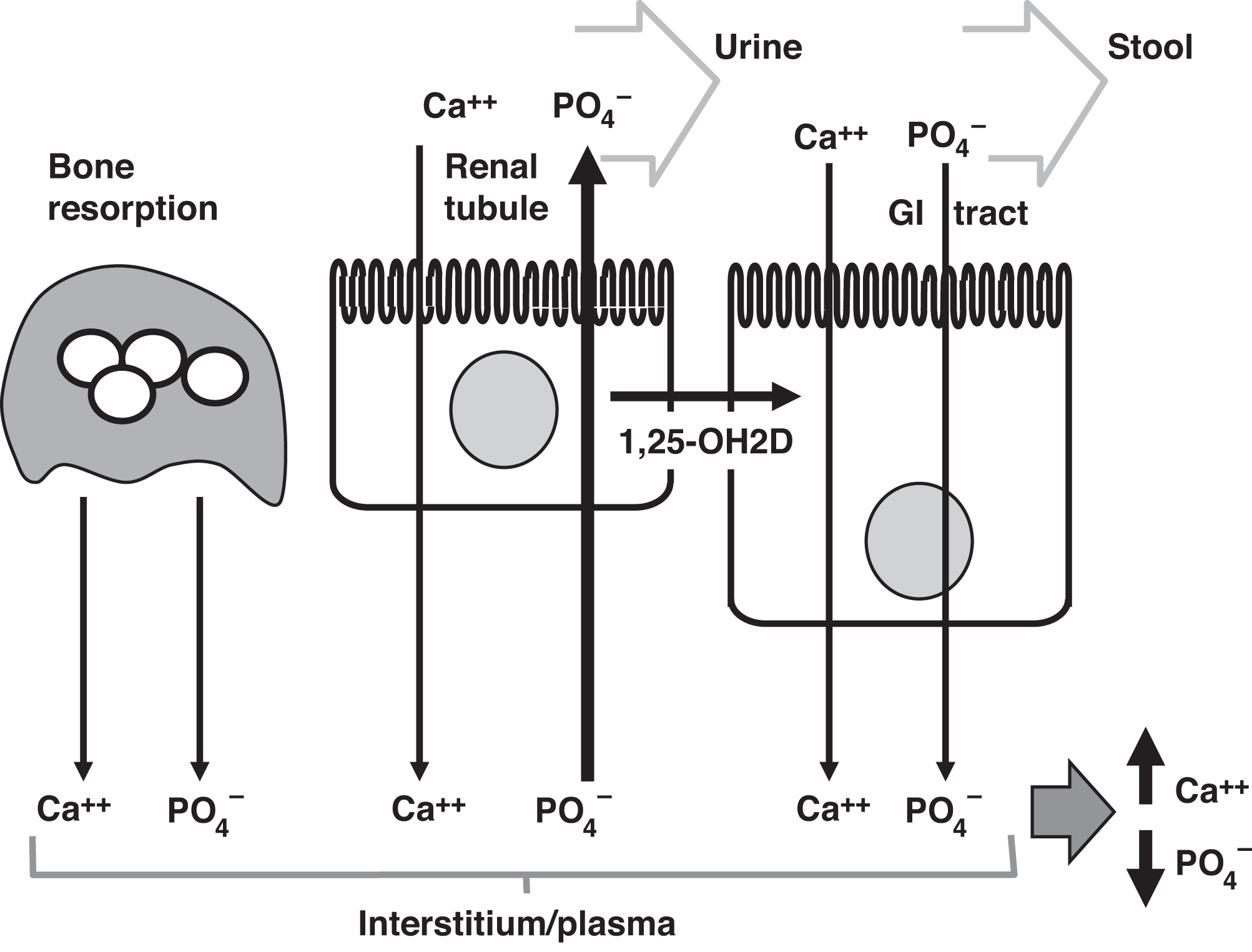
In summary, elevated PTH enhances bone resorption and increases calcium and phosphate concentrations in the blood due to bone resorption. Increased absorption of calcium and phosphate from the intestine stimulated by 1,25-OH2D (consequent to an elevated PTH action) also leads to increased calcium and phosphate in plasma. PTH does increase renal tubular calcium reabsorption to increase calcium concentration. However, PTH causes phosphaturia that is of sufficient magnitude to reduce circulating phosphate concentrations despite its effects on the intestine and bone in raising phosphate. Thus, the overall effect of PTH is to raise calcium and decrease phosphate concentrations ( Fig. 10.15 ). From this basic physiology, it can be readily predicted that PTH excess can cause hypercalcemia, hypophosphatemia, and an elevated alkaline phosphatase concentration, whereas PTH deficiency causes hypocalcemia and hyperphosphatemia.
PO 4 3 −
). PTH increases renal tubular absorption of calcium and increases phosphaturia serving to lower plasma phosphate. PTH stimulates the conversion of 25-OHD to 1,25-OH2D in the kidney. 1,25-OH2D stimulates increased absorption of calcium and phosphate from the GI tract. Overall, PTH raises plasma calcium concentrations and lowers plasma phosphate concentrations.
If the plasma calcium and phosphate concentrations are both elevated to the higher reference intervals or above, ectopic calcification can occur. As a rule of thumb, if the calcium concentration (in mg/dL) is multiplied by the phosphate concentration (in mg/dL) and exceeds ~70, ectopic calcification is possible. Sites of calcium deposition can include the renal tubules, skin, and vasculature. The mechanisms of ectopic calcification are still debated but may involve matrix vesicle secretion .
Parathyroid hormone assays
The measurement of PTH deserves special attention because of its diagnostic and prognostic importance. In chronic kidney disease, higher PTH levels correlate with greater degrees of metabolic bone disease.
First-generation PTH assays of the 1960s and 1970s were polyclonal competitive radioimmunoassays with wide specificity for PTH and PTH-related fragments. Second- and third-generation PTH immunoassays are two-site, noncompetitive (“sandwich”) assays. These methods have not been harmonized . The two-site assays were originally termed “intact PTH” assays (vs the older mid-region, C-terminal, or N-terminal PTH competitive radioimmunoassays) before it was appreciated that they measure both intact hormone (PTH[1–84]) and N-terminal-truncated fragments such as PTH(7–84). Detection of PTH(1–84) and PTH(7–84) results from an N-terminal-detecting antibody that does not require the presence of the first 6 N-terminal amino acids in the PTH molecule. These two-site assays that detect (PTH[1–84]) and predominantly PTH(7–84) constitute the second-generation PTH assays developed in the 1980s.
Several diagnostics companies have since developed immunoassays that measure intact PTH(1–84) but not PTH(7–84). These third-generation PTH assays have also been termed “biointact,” “whole,” “bioactive,” or “cyclase-activating” PTH (CA-PTH) PTH assays. Cyclase activating refers to the ability of PTH(1–84) to stimulate adenylate cyclase. There is evidence that a larger “intact” PTH fragment is also detected by these assays .
There was concern that because the intact PTH assays measure PTH(7–84) and other N-terminal-truncated forms in addition to PTH(1–84) that these immunoassays could overestimate PTH concentrations in patients with renal failure and secondary hyperparathyroidism (see below). The worry was then that if the PTH was overestimated, vitamin D treatment (e.g., calcitriol) of secondary hyperparathyroidism would be overly aggressive, possibly leading to vitamin D toxicity. If one compares the concentrations of PTH measured by the intact PTH assays and the biointact PTH assays, although the results are significantly lower (slope=0.52) with biointact PTH assays, the two methods are highly correlated ( r =0.985) . This emphasizes the importance of establishing separate target ranges for intact PTH versus biointact PTH concentrations that are method-specific because of the lack of PTH measurement harmonization. That being said, intact PTH and biointact PTH are highly correlated whether the patients are on dialysis, are uremic, or have primary hyperparathyroidism . With these results in hand, the biointact PTH assay has yet to significantly alter clinical practice .
In one informative study, biointact PTH results did not correlate better with markers of bone turnover than intact PTH results . Also, if the ratio of PTH(1–84) to PTH(7–84) is calculated, this ratio does not correlate with the rate of bone formation (24). In receiver–operator characteristic curve evaluation of the ability of PTH to predict bone turnover, intact PTH and biointact PTH give similar areas under the curve (AUCs), whereas the ratio of biointact PTH to intact PTH did not perform as well as either marker alone. This is also true in uremic patients where the ratio assessment performs even more poorly .
Individual second-generation assays exhibit good PTH reproducibility (e.g., coefficients of variation of <10%). Because of the lack of PTH assay harmonization, differences between methods usually exceed 20%. The International Federation of Clinical Chemistry (IFCC) is working to harmonize PTH measurements . A 2005 study reported a reference interval for intact PTH measuring PTH(1–84) and PTH(7–84)] of 10–65 pg/mL for “normal” subjects and 10–46 pg/mL for vitamin D-sufficient subjects .
Clinical manifestations of disordered calcium or phosphate metabolism
Disorders of calcium metabolism clinically present as hypocalcemia, hypercalcemia, bone disease (e.g., rickets), and/or renal disease (e.g., impaired renal function). Patients with hypocalcemia can experience muscle cramps, muscle spasms, paresthesias, tetany, or seizures. Electrophysiologically, hypocalcemia affects the sodium channel by lowering its threshold for depolarization. Hypocalcemia can prolong the patient’s Q–T interval on electrocardiographic (EKG) testing, even causing arrhythmias with severe hypocalcemia.
The spectrum of inherited bone disorders is very wide . Many of these disorders involve mutations in collagen , various receptors , SHOX (short stature homeobox; chromosome Xp22.33) , and the ALPL gene (alkaline phosphatase, biomineralization associated; chromosome 1p36.12) as prime examples. The 11th International Skeletal Dysplasia Society describes 42 unique forms of skeletal dysplasias resulting from mutations in 346 genes !
Supportive clinical evidence of hypocalcemia includes demonstration of the Chvostek sign and the Trousseau sign. The Chvostek sign is ipsilateral (e.g., same side) facial contraction when the facial nerve is lightly tapped with the examiner’s fingers. The Trousseau sign is forearm muscle contract with brachial artery occlusion for 3 min using a blood pressure cuff (e.g., the cuff pressure is 10 mmHg above the systolic pressure). Complications of chronic hypocalcemia include basal ganglia calcification and cataract formation.
Mild hypercalcemia (e.g., total calcium <12 mg/dL) may be asymptomatic. Modest hypercalcemia can produce vague complaints of fatigue, malaise, weakness, depression, apathy, and an inability to concentrate. Chronic hypercalcemia can impair the concentrating ability of the kidney (e.g., nephrogenic diabetes insipidus). Patients with hyperparathyroidism classically complain of psychic disorders, renal stones (nephrolithiasis ) or nephrocalcinosis (i.e., in the kidney parenchyma the ectopic deposition of calcium salts ), GI disorders, and bone pain (e.g., osteopenia, cystic bone lesions, pathologic fractures). Subperiosteal bone resorptions noted in the fingers and around the teeth are classic radiologic findings in hyperparathyroidism.
Hypocalcemia
A reduced level of total calcium in the blood requires confirmation by measurement of either albumin (in search of hypoalbuminemia) or ionized calcium (the better approach). If the reduction in calcium is not due to reduced carrier protein concentrations (e.g., hypoalbuminemia), “true” hypocalcemia is present. There are numerous causes for hypocalcemia ( Table 10.5 ) . Clinically, the most prevalent chronic calcium disorders occur in patients with renal failure. Renal failure will be discussed below.
|
Decreased parathyroid hormone action
Reduced PTH activity can result from hypoparathyroidism or PTH resistance (e.g., pseudohypoparathyroidism). Reduced PTH activity is manifested as hypocalcemia and hyperphosphatemia. Alkaline phosphatase concentrations are not elevated because PTH action is deficient. In hypoparathyroidism, the PTH level is inappropriately low for the ionized calcium concentration. The PTH concentration may be below the reference interval or within the reference interval (which is still inappropriate in a hypocalcemic patient). In cases of PTH resistance, the PTH is elevated because of PTH resistance, and results in hypocalcemia and hyperphosphatemia. The symptoms of hypocalcemia are as described above.
Parathyroid hormone deficiency—acquired hypoparathyroidism
Hypoparathyroidism may exist as an isolated disorder or may be associated with other endocrine or nonendocrine disorders. Isolated hypoparathyroidism can be idiopathic or autoimmune. Rarely, the parathyroid glands are destroyed by cancer metastases, iron deposition (e.g., hemochromatosis) , or copper deposition (e.g., Wilson disease) . Acquired causes of hypoparathyroidism are listed in Table 10.6 .
|
Autoimmune hypoparathyroidism can occur sporadically or as part of a genetic autoimmune polyglandular syndrome (APS) (see below) . Currently, there are no clinically available tests for parathyroid autoantibodies. However, in research studies, autoantibodies to NAIP, CIITA, HET-E, and TP1 (NACHT) leucine-rich-repeat protein 5 (NALP5) and the CaSR have been described. CaSR-specific cytotoxic CD8 T cells have been reported in hypoparathyroidism . Immune checkpoint inhibitor therapy has caused antibody-mediated hypoparathyroidism .
During thyroidectomy, unintended parathyroidectomy may occur . Irradiation can damage the parathyroid glands . Transient hypoparathyroidism in the setting of acute severe illness is common in infants, children, and adults. This is especially true in premature infants, sick term newborns, and infants of diabetic mothers . Vitamin D deficiency and hypomagnesemia can contribute to hypocalcemia in newborns . Maternal hypercalcemia or maternal normocalcemic hyperparathyroidism can transiently suppress the parathyroid glands of their newborns (because of presumed in utero hypercalcemia), leading to transient hypocalcemia .
Parathyroid hormone deficiency—genetic causes of hypoparathyroidism
Hypoparathyroidism is observed in several genetic conditions as listed in Table 10.7 . If there is a gain-of-function mutation in the CaSR (a type of “receptoropathy”), the parathyroid glands are overly sensitive to calcium, and there is decreased PTH release causing hypocalcemia and hyperphosphatemia . This disorder is also known as “autosomal-dominant hypocalcemia with hypercalciuria type 1 (ADH1)” . Autosomal-dominant hypocalcemia with hypercalciuria type 2 (ADH2) is a consequence of GNA11 (G protein subunit α 11; chromosome 19p13.3) gain-of-function mutations . GNA11 is a G-protein subunit α involved in CaSR signaling in the parathyroid chief cells. The gain-of-function mutation is similar to the CaSR gain-of-function mutation where the parathyroid gland senses increased calcium concentrations and lowering PTH secretion despite the fact that the plasma calcium concentration is really not elevated.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


