Chapter 6 Didanosine
STRUCTURE
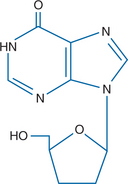
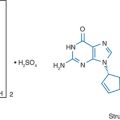
Didanosine (2′,3′-dideoxyinosine, ddI, Videx, Videx EC) is a nucleoside analog active against human immunodeficiency virus type 1 (HIV-1), HIV-2, simian immunodeficiency virus, and human T-cell lymphotropic virus-1 (HTLV-1)1,2 (Fig. 6-1). It is acid-labile and under acidic conditions is hydrolyzed tonn 2′,3′-dideoxyribose and the base hypoxanthine.3 Didanosine is also the deamination product in vivo of 2′,3′-dideoxyadenosine (ddA) through the action of the ubiquitous enzyme adenosine deaminase.4 Didanosine was the second antiretroviral drug approved by the US Food and Drug Administration (FDA) in 1991.
MECHANISM OF ACTION
Didanosine exerts its antiviral action through its triphosphorylated metabolite 2′,3′-dideoxy-adenosine 5′-triphosphate (ddATP), which competes with the naturally occurring nucleoside 2′-deoxyadenosine 5′-triphosphate (dATP) for binding to HIV-1 reverse transcriptase (RT).5 In addition, ddATP serves as a chain terminator after incorporation into viral DNA because it lacks the requisite 3′-hydroxyl group for elongation of the DNA nascent chain.6,7
Didanosine enters the cell by means of nonfacilitated diffusion through a nucleobase carrier.8 It is initially phosphorylated to the monophosphate form (ddIMP) by a 5′-nucleotidase and then converted to ddA monophosphate by an adenylosuccinate synthetase and adenylosuccinate lyase.3 It is subsequently phosphorylated to the diphosphate (ddADP) and finally the triphosphate (ddATP), which is the active antiviral moiety. ddATP has a prolonged intracellular half-life, estimated to be 8–40 h.9,10 This provides the rationale for the prolonged dosing intervals of once or twice per day for didanosine used in clinical trials.
Didanosine is active against HIV-1 in both lymphocytes and macrophages, with a median inhibitory concentration (IC50) of 0.24–0.6 mg/L in T-cell cultures and 0.002–0.020 mg/L in monocyte/macrophage cultures.6,11–13 Didanosine appears to have more activity in resting cells than in activated cells, in contrast to zidovudine or stavudine for which the reverse is true.14,15 The antiviral activity of didanosine also appears to be relatively unaffected by the presence of endogenous nucleosides such as 2′-deoxyadenosine.3 In vitro, didanosine almost completely inhibits HIV replication at concentrations of 4.8 mg/L.16 Against HIV-1, didanosine demonstrates additive effects or synergy with nucleoside analogs such as zidovudine and stavudine,17 non-nucleosidereverse transcriptase inhibitors (NNRTIs) such as delavirdine,18 and protease inhibitors (PIs) such as saquinavir18 or indinavir.19 Hydroxyurea demonstrates a synergistic effect against HIV-1 when combined with didanosine through inhibition of the ribonucleotide reductase enzyme. This results in the decrease of intracellular dATP with which the active metabolite of didanosine (ddATP) competes for binding to RT.20 Didanosine is approximately 100-fold less toxic than zidovudine against bone marrow progenitor cells,16 which likely accounts for the lack of hematopoietic toxicity of the drug observed in clinical trials.
PHARMACOKINETICS
The N-glycocidic bond of didanosine is highly acid-labile, and significant degradation occurs at the low pH found in gastric fluid.21 Thus, oral preparations have been formulated with buffers to prevent degradation or as an enteric coated delayed release preparation (Videx EC). The sachet (powder) form contains citrate phosphate buffer, and the chewable/dispersible tablet contains calcium carbonate and magnesium hydroxide as antacids. A pediatric powder formulation of didanosine is also available without buffer and must be reconstituted with water and administered with a concomitant antacid. The nonbuffered, enteric coated, delayed-release formulation consists of a capsule with enteric coated beadlets that contain didanosine (see further ahead).22
Oral bioavailability of didanosine has been reported to be somewhat variable and dose dependent.3,23 The buffered chewable/dispersible tablet is 20–25% more bioavailable than the sachet form, which accounts for the lower recommended daily dose of the tablet (usually 400 mg/day for patients weighing 60 kg or more) compared to the powder (500 mg/day).4,10 Twice-a-day dosing of the buffered tablet was the regimen initially recommended and studied most extensively. However, once-daily administration of twice the dose given BID results in similar plasma concentration–time curves (area under the curve, or AUC),24 as does administration of the enteric coated delayed-release formulation, which should be administrated only once a day (Table 6-1).22 Once-a-day dosing has been approved by the FDA, and is now used most frequently, although the FDA states in the package insert that twice-a-day dosing is preferred because “there is more evidence to support its effects” (see further ahead). The bioavailability of didanosine is decreased up to 50% when administered with food and so should be taken on an empty stomach, either 30 min before or 2 h after a meal.22,25,26

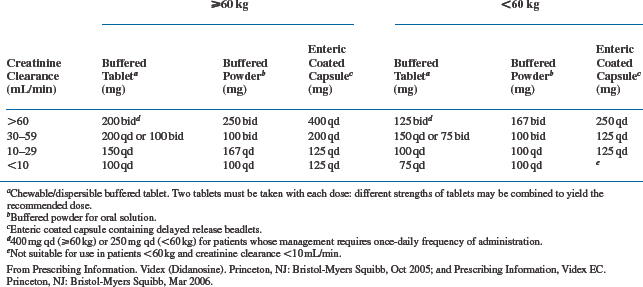
Oral absorption of buffered didanosine preparations is generally rapid, and time to peak plasma concentrations (Tmax) occurs 0.50–1.13 h after oral administration.3,6,7,27 Peak concentrations range between 5 and 9 μmol/L when currently recommended doses are used.3 The peak concentration for the EC delayed-release preparation is ∼40% lower, and Tmax is prolonged to 2 h.22 Approximately 5% of didanosine is bound to protein.16 The plasma elimination half-life of didanosine after administration is relatively short, ranging from 0.50 to 2.74 h after multiple oral doses.6,7 Thirty tosixty percent of an oral dose is excreted unchanged by the kidneys via glomerular filtration and tubular secretion.23,28 Reduction of dosage is recommended for patients with significant renal dysfunction (creatinine clearance is less than 60 mL/min), as shown in Table 6-2.10,22 Didanosine is partially removed during hemodialysis, with an extraction ratio of 53%.29 For patients requiring hemodialysis or continuous ambulatory peritoneal dialysis (CAPD), didanosine buffered or EC formulations should be given the dose which is appropriate for those with a creatinine clearance of less than 10 mL/min.10,22
As noted above, didanosine is converted to the active moiety ddATP and then further metabolized to hypoxanthine and eventually to uric acid. Didanosine is converted to hypoxanthine by a purine nucleoside phosphorylase (PNP), which is also inhibited by tenofovir (TDF). This apparently accounts for the increased concentrations of ddI when administered with TDF,30 and was the basis for attempts to use combinations of TDF and ddI to avoid dietary restrictions with the latter.31,32 However, the ddI–TDF combination has resulted in increased rates of toxicity (see further ahead). The volume of distribution of didanosine is 0.76–1.29 L/kg,33 which is less than the distribution of zidovudine, likely reflecting the lower lipid solubility of didanosine. Mean cerebrospinal fluid concentrations have been reported to be 21% of serum concentrations (range 12–85%), although the concentrations were more variable in children.28,33,34
Pharmacokinetic measurements in pregnant patients were not significantly different from those obtained in nonpregnant patients.35 Didanosine was found in placental and fetal circulations at concentrations that were 20–50% of those in the maternal circulation, presumably because of placental metabolism of the drug.35–37 The pharmacokinetics of didanosine in pediatric patients are similar to those described in adults, although bioavailability was somewhat lower in some studies of children.38–40 Once-daily dosing of didanosine (180 mg/m2) in pediatric patients had similar bioavailability (90 mg/m2) when given twice-daily.41
TOXICITY
The currently available formulations of didanosine, including the dispersible buffered tablet and the extended-release capsule, are more palatable than earlier formulations. This, along with once-daily dosing, has improved acceptability of the drug. Overall, didanosine has been a reasonably well tolerated drug as ascertained by large-scale clinical studies of HIV-infected patients at various stages of disease progression.42–44 However, significant toxicities may be encountered, of which the most common are listed in Table 6-3. The most well-established side effects are peripheral neuropathy and pancreatitis, which have also been the dose-limiting toxicities encountered in phase I dose-escalation studies.44 Adverse effects occur most frequently in patients with advanced HIV disease and do not appear to differ significantly between once-a-day and twice-a-day regimens of didanosine, except for rates of diarrhea, which may be decreased with the EC formulation.10,22

Peripheral neuropathy associated with didanosine is indistinguishable from that seen with other nucleoside analogs such as zalcitabine. It is primarily a sensory polyneuropathy that is usually symmetrical and distal; and it most commonly involves the lower extremities.45–47 Initial symptoms are often dysesthesias, which are described as aching, burning, tingling or numbness. They can progress to markedly disabling pain on ambulation or even at rest. Phase I studies indicated that the frequency of neuropathy was dose-related and decreased when doses below 12 mg/kg/day were used.45 In the expanded access program in which patients with advanced HIV disease received didanosine, the reported frequency of peripheral neuropathy was 16%,10 which undoubtedly includes a background of neuropathy caused by other drugs and by underlying HIV infection. In two large-scale clinical trials that compared didanosine and zidovudine monotherapy (AIDS Clinical Trial Group (ACTG) 116a42 and ACTG 116b/11743), the rates of peripheral neuropathy were not significantly different between the didanosine- and zidovudine-treated groups. Because zidovudine is not known to cause peripheral neuropathy, these studies indicate that peripheral neuropathy caused by didanosine is uncommonly encountered when recommended doses of the drug are employed. Nonetheless, clinicians must be aware of risk factors for the development of neuropathy with didanosine, which include the presence of underlying peripheral neuropathy from other causes, concurrent treatment with other potentially neurotoxic drugs,48 and low CD4+ T-lymphocyte counts.49 Rates of peripheral neuropathy appear to be increased when didanosine is combined with hydroxyurea or with stavudine and probably with TDF (see further ahead).50,51 Peripheral neuropathy associated with didanosine generally resolves over several weeks after discontinuing the drug, although occasionally symptoms worsen for 1–2 weeks after the drug has been stopped. Some patients with didanosine-associated peripheral neuropathy have been able to tolerate reinstitution of the drug at lower doses,52 but this is rarely necessary in an era when multiple alternative antiretroviral therapies are available. The pathogenesis of nucleoside-induced peripheral neuropathy is not fully understood but appears to be related to inhibition of mitochondrial DNA polymerases in neurons by this class of drugs.53
Acute pancreatitis is a dose-related toxic effect of didanosine that is infrequently encountered at currently recommended doses.54 As is the case with peripheral neuropathy, pancreatitis unrelated to didanosine can develop in HIV-infected patients because of opportunistic infections or neoplasms that involve the pancreas and the use of other pancreatotoxic drugs.55 Among 7806 patients who participated in the expanded access program for didanosine, pancreatitis was noted in 5% who received the drug for at least 5 months.10 Acute pancreatitis associated with didanosine can present with variable severity, ranging from mild abdominal pain to life-threatening illness on rare occasions.54,56 Fatalities associated with pancreatitis occurred in 0.35% of patients in the expanded access program.10 Asymptomatic elevations in serum amylase and, to a lesser extent, lipase and triglycerides have also been observed.3,57 In the large-scale studies that compared zidovudine and didanosine monotherapy noted above (ACTG 116a42 and ACTG 116b/11743), an excessive rate of pancreatitis (3–4%) was noted in the didanosine arm (500 mg/day) compared to the zidovudine monotherapy arm. In the ACTG 175 trial, which examined patients with somewhat less advanced HIV disease, the overall frequency of pancreatitis was 0.5% in the entire study population and was not increased in the didanosine-treated groups.58 Risk factors for the development of pancreatitis include a history of pancreatitis, a history of alcohol or drug abuse, hypertriglyceridemia, and possibly renal dysfunction.10,59 Co-administration with TDF also increases risk of pancreatitis (see further ahead).31 Combination of didanosine with stavudine, with or without hydroxyurea, may also increase the risk of pancreatitis.10,22,60 Clinicians who treat patients with didanosine should be alert to signs and symptoms of pancreatitis, and the drug should be stopped promptly if pancreatitis is suspected. Some patients with risk factors for the development of pancreatitis may benefit from periodic monitoring of amylase, lipase, and triglycerides. If amylase concentrations rise 1.5–2.0 times above the upper limit of normal or if triglycerides rise above 7 g/L, didanosine administration should be stopped.48 Fractionation of serum amylase into total and salivary components may increase the specificity of such monitoring. Although some patients with didanosine-associated pancreatitis have been able to tolerate lower doses of the drug once the acute episode has resolved,45,48 didanosine should be avoided in such patients unless there is no other alternative for antiretroviral therapy (ART).
Diarrhea has also been associated with didanosine administration. It has been attributed, at least in part, to the citrate phosphate buffer in the powdered preparation and may be less frequent with the EC formulation.22 Diarrhea was reported in 16% of patients in the didanosine expanded-access program in which patients had a far advanced HIV disease and were receiving multiple other medications.61 In studies of monotherapy with didanosine in which the chewable/dispersible tablet was employed, rates of diarrhea were not significantly different from those seen with zidovudine monotherapy arms in some studies. Elevations in aminotransferases (more than five times normal) were seen in 6–9% of subjects who received didanosine monotherapy,10,42,43 and rare cases of fulminant hepatitis have been reported.62,63
As with other nucleoside analogs, life-threatening lactic acidosis and hepatic steatosis may occur with didanosine.64–66 The FDA and the manufacturers of didanosine issued a letter of warning reporting three fatal cases of lactic acidosis in pregnant women who were receiving didanosine combined with stavudine and recommended that this combination be used in pregnant women only when the ‘potential benefit clearly outweighs the risk.’67 Asymptomatic elevations in uric acid have also been noted, primarily in patients with preexisting hyperuricemia.61 Rarely, depigmented retinal lesions and optic neuritis have been reported in patients receiving didanosine,68,69 and consideration of periodic retinal examinations for patients on didanosine have been suggested.10,22,70
Of particular note has been the absence of hematopoietic toxicity associated with didanosine administration. In fact, in several studies, improvement in hemoglobin, white blood cells, and platelet counts occurred on didanosine therapy, presumably because of the beneficial effects of the drug in inhibiting replication of HIV.4,71
The recommended dosage schedule for didanosine was twice a day for the buffered formulation, but in 1999 the FDA approved once-a-day dosing,10 as well as the EC delayed-release formulation, which should be administered only once a day.22 Comparative trials of twice- and once-a-day regimens of didanosine in combination with stavudine have demonstrated equivalent effects on virologic and CD4 cell count parameters,72,73 and similar findings were noted when twice- and once-a-day regimens of didanosine were used in combination with zidovudine.74 Once-a-day dosing of didanosine, in combination with stavudine and nelfinavir, had an antiretroviral effect equivalent to that of a combination of zidovudine, lamivudine, and nelfinavir.10 Buffered didanosine preparations administered on a once-a-day regimen has also been employed in other comparative and noncomparative studies of didanosine in a wide variety of combination trials, including nucleoside reverse transcriptase inhibitors (NRTIs), NNRTIs, and PIs.7,75–78 The FDA currently points out that for adults, ‘the preferred dosing frequency of VIDEX (ddI chewable/dispersible buffered tablets) is twice daily because there is more evidence to support the effectiveness of this dosing regiment.’10 Once-daily dosing should be considered only for ‘adults whose management requires once-daily administration of Videx.’10 Videx EC (delayed release capsules containing enteric coated beadlets) should be administered once daily.22
RESISTANCE
After didanosine monotherapy, five- to 26-fold reduction in didanosine sensitivity has been reported; it is associated with a change in the pol gene at codon 74 (Leu:Val), which appears to be the predominant mutation associated with resistance.13 Isolates from 60% to 65% of patients who have received didanosine therapy for at least 1 year demonstrate this mutation.79–81 Viruses with the L74V mutation also manifested decreased virus replication (‘fitness’) in vitro.82 A similar level of resistance with a change at codon 184 (Met:Val) has been reported.83,84 A mutation at codon 65 (Lys:Arg) has resulted in a three- to fivefold decrease in sensitivity85; and mutations at 135 (Ile:Val) and 200 (Thr:Ala), which confer resistance, have also been described.86
Mutations at codon 74 are also associated with partial restoration of susceptibility to zidovudine but with an approximately 10-fold reduction in sensitivity to zalcitabine.12,87 The position K65R mutation is associated with a reduction insensitivity to both zalcitabine and lamivudine,88 and appeared to have an added effect on resistance to didanosine when present with the M184V mutation.89 The development of a mutation at codon 41, 67, 70, 215, or 219 conferred resistance to zidovudine in patients who received prolonged didanosine therapy but apparently had not received zidovudine therapy.90 This suggests that viruses resistant to both zidovudine and didanosine occasionally emerge in patients on didanosine therapy alone. Resistance mutations at codon 74 have been associated with poorer CD4 responses and higher virus loads in patients receiving didanosine.81
Combination therapy with zidovudine and didanosine reduced the rate of emergence of genotypic didanosine resistance to less than 4% compared to patients who received didanosine monotherapy.79,80,91–93 Combination therapy with didanosine and zidovudine did not decrease the emergence of viruses resistant to zidovudine in studies that examined the sensitivity of isolates to both drugs.91,94
Viruses resistant to both didanosine and zidovudine have been detected after prolonged combination therapy with both drugs, with amino acid substitutions at codons 62, 75, 77, 116, and 151.91,95–97 These viruses are also resistant to zalcitabine and stavudine and are partially resistant to lamivudine. The inclusion of the mutation Glu to Met at codon 151 (Q151M) and three or four other mutations that result in broad NRTI resistance have been associated with loss of favorable effect on CD4+ T-lymphocyte counts.98–99 A 6-bp insert between codons 69 and 70 that confers broad resistance to NRTIs has also been identified in patients who received didanosine or zalcitabine alone or in combination with zidovudine.100
DRUG INTERACTIONS
Drug interactions that occur with the buffered formulations of didanosine are most frequently related to the antacid activity in the tablet or powder or to the divalent calcium and magnesium cations present in the tablet form or in antacids added to the pediatric powder (Table 6-4). These interactions can be minimized by administering other drugs 2 h before or 2–6 h after administration of didanosine. An example of this type of interaction is the reduction in the absorption of drugs such as itraconazole101,102 or ketoconazole,103,104 which require gastric acidity for optimal absorption when these drugs are given concomitantly with didanosine. Dapsone also requires low gastric pH for optimal absorption, and there was concern that buffering by didanosine might account for the apparent failure of dapsone prophylaxis for Pneumocystis jerovici infection when both drugs were administered to patients.105 However, pharmacokinetic studies in humans receiving didanosine did not show a reduction of plasma dapsone levels.10,22,106 Absorption of tetracycline107 and ciprofloxacin108 (and possibly other quinolone antibiotics) is decreased by chelation with divalent cations in didanosine tablets. Concurrent administration with ranitidine and presumably other H2-blockers results in a small increase in bioavailability of buffered didanosine preparations by decreasing gastric acidity.109 Concomitant ganciclovir administration increases plasma didanosine concentrations by 57.7% (Cmax) and AUC values by 71.5%,110–112 so clinicians should be alert to the possibility of increased didanosine toxicity when the two drugs are given together. Studies of interactions between didanosine and zidovudine have shown only minor changes in the pharmacokinetics of either drug.113–115 When delavirdine and didanosine were administered simultaneously, reductions in the concentrations of both delavirdine (32% decrease in AUC) and didanosine (20% decrease in AUC) were noted116,117; hence the two drugs should be given at least 1 h apart. Decreased absorption of indinavir has also been noted when the drug is administered simultaneously with didanosine,116,118 but not if didanosine is taken 1 h before indinavir.119 Minor interactions, without clinical significance, have been observed between didanosine and ritonavir.120 Clinically significant pharmacokinetic interactions have not been observed between didanosine and the following drugs: trimethoprim-sulfamethoxazole,121 rifabutin,122 isoniazid,123 foscarnet,124 stavudine,125 loperamide,22 metoclopramide,22 clarithromycin,126 nevirapine,127 and nelfinavir.128 Administration of allopurinol increases the AUC of didanosine approximately fourfold through inhibition of the enzyme, PNP, so the two drugs should not be administered together.10,22 Chronic methadone administration reduces the AUC of didanosine by 57% and the Cmax by 66%.129
Table 6-4 Drug Interactions with Didanosine
| Type of Interaction | Drug | Refs |
|---|---|---|
| Decreased absorption of drugs requiring gastric acidity for optimal absorptiona | Itraconazole | 101, 102 |
| Ketoconazole | 103, 104 | |
| Decreased absorption of drugs by chelation with cationsa | Tetracycline | 107 |
| Ciprofloxacin | 108 | |
| Increased absorption of didanosine by block of gastric acid production | Ranitidine | 109 |
| Increased plasma concentration of didanosine | Ganciclovir | 110–112 |
| Decreased plasma concentration of drug when given at the same time as didanosineb | Delavirdine | 116, 117 |
| Indinavir | 116, 118 | |
| Decreased plasma concentration of didanosine when given with drug | Delavirdine | 116, 117 |
| Methadone | 118 | |
| Possibly increased rate of peripheral neuropathy when given with didanosine | Isoniazid | 48 |
| Ethambutol | ||
| Metronidazole | ||
| Zalcitabine | ||
| Hydroxyurea | 50, 51 | |
| Stavudine | ||
| Possibly increased pancreatic toxicity when given with didanosine | Pentamidine | 55 |
| Azathioprine | ||
| Ethambutol | ||
| Zalcitabine | ||
| Stavudine | 60 | |
| Increased plasma concentration of didanosine through inhibition of PNP | Allopurinol | 10, 30 |
| Tenofovir | 130, 131 |
a Interaction is avoided if a nonbuffered preparation (VIDEX EC) is used.
b Interaction is avoided if didanosine is administered 1–2 h before indinavir or 1 h apart from delavirdine.
Administration of TDF may increase the plasma levels of didanosine by 40% or more,130,131 through inhibition of PNP. This likely accounts for the increased toxicity observed in co-administration of TDF and didanosine, particularly as increased rates of pancreatitis,132 and possibly peripheral neuropathy as well. An interaction between didanosine and TDF resulting in enhanced mitochondrial toxicity has been suggested as well.133 Increased rates of hyperglycemia have also been reported in combination therapy with the two drugs.134 Because of the above toxicities, and apparent decreased efficacies (see further ahead), concomitant use of TDF and didanosine should be avoided if possible. If they need to be used together, the manufacturers suggest that the dosage of didanosine be reduced to 250 mg daily for adults weighing 60 kg or greater, or 200 mg for adults weighing under 60 kg with creatinine clearances of 60 mL/min.10,22 The EC formulation of didanosine, which does not contain buffer, should not be expected to result in drug interactions that depend on the antacid or cation contents of didanosine tablets. However, drug interactions with didanosine based on other mechanisms, such as those involving allopurinol, ganciclovir, or methadone, would be expected to occur with the EC formulation as well.22
As noted in the section on toxicity, clinicians should be alert to the development of didanosine toxicity when drugs that are known to cause or potentiate peripheral neuropathy49 (e.g., isoniazid, ethambutol, metronidazole, vincristine, hydroxyurea, stavudine and possibly TDF) or pancreatitis54,60 (pentamidine, azathioprine, TDF, stavudine) are co-administered, and alternative drugs should be considered. Because of the potential for additive toxicities involving peripheral neuropathy and pancreatitis, didanosine and zalcitabine should not be administered concurrently.
EFFICACY
Didanosine Monotherapy or Dual Nucleoside Combination Therapy
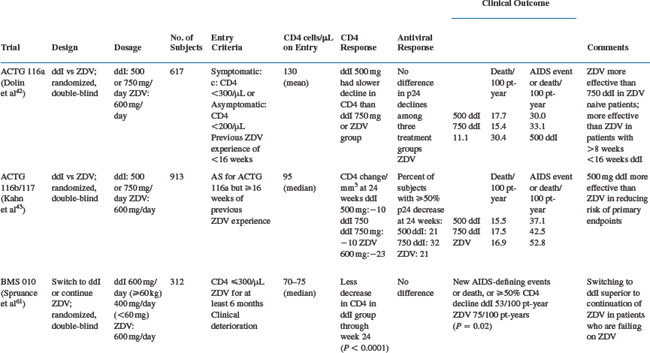
Clinical studies of didanosine monotherapy or dual combination therapy were carried out at times when the use of one or two antiretrovirals was the standard of care. The major studies of this type are summarized in Table 6-5. Studies conducted by the ACTG showed that changing to didanosine monotherapy was more effective than continuing zidovudine monotherapy in patients with advanced HIV infection who had previously received at least 8 weeks of zidovudine (ACTG 116a, ACTG 116b/117),42,43 although zidovudine appeared to be somewhat more effective than didanosine in antiretroviral-naive patients.42 Studies of patients who received at least 6 months of zidovudine and were either clinically stable135 or deteriorating61 also showed benefits after their therapy was changed to didanosine. A meta-analysis of the above studies and ACTG 175 (see further ahead) supported the beneficial effect of switching from zidovudine to didanosine across a broad range of CD4 and HIV disease categories.136 A comparison of didanosine with zidovudine as initial monotherapy in patients with ‘mildly symptomatic disease’ and CD4 counts lower than 500/μL showed that the clinical efficacies of the two drugs were similar.137



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


