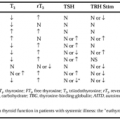
DIAGNOSIS
POSTNATAL DIAGNOSIS
The diagnosis of 21-hydroxylase deficiency may be confirmed by administering an intravenous bolus of ACTH and measuring the resultant elevation in blood levels of 17-hydroxyprogesterone.26 Usually, a panel of adrenal hormones is assayed before and after ACTH administration, but the most specific available marker for which testing is commercially available is 17-hydroxyprogesterone. Clinicians should be aware that cortisol stimulation is suboptimal after ACTH infusion in patients with severe defects in adrenal steroid synthesis. If for any reason blood testing cannot be used or radioimmunoassays for 17-hydroxyprogesterone are unavailable, the examiner can measure 17-ketosteroids or pregnanetriol in a 24-hour urine collection. The latter steroid is the principal direct urinary metabolite of 17-hydroxyprogesterone.
Ancillary tests used in the initial evaluation of infants with ambiguous genitalia include karyotype analysis, pelvic and abdominal ultrasonography, and sonogram of the urogenital orifices using radiopaque dyes.
Patients with nonclassic 21-hydroxylase deficiency have 17-hydroxyprogesterone levels that exceed those seen in heterozygous carriers of an affected gene, but these levels are lower than those of patients with the classic form of the disorder.26 In the nonstimulated state, these patients may have near-normal serum hormone levels. A serum 17-hydroxyprogesterone level below 200 ng/dL effectively excludes this diagnosis if the sample is obtained in the early morning (i.e., by 8:00 a.m.).
The diagnosis of 11β-hydroxylase deficiency is made by the measurement of elevated basal or ACTH-stimulated DOC and/or 11-deoxycortisol (i.e., compound S) in the serum or elevated levels of the tetrahydro-compounds (i.e., DOC and/or S) in a 24-hour urine collection.27 Another marker useful in pediatric diagnosis is 6α-hydroxytetrahydro-11-deoxycortisol, which can be measured by gas chromatography and mass spectrometry of urine.28 As in 21-hydroxylase deficiency, urinary 17-ketosteroids are usually elevated, reflecting increased shunting of 11β-hydroxylase hormonal precursors into the sex steroid pathway. PRA is usually low in older children and is accompanied by low levels of aldosterone.
The diagnosis of 17α-hydroxylase/17,20-lyase deficiency is made by a finding of marked elevations of serum DOC and corticosterone
(i.e., compound B) and the metabolites of these two steroids.14 Aldosterone is often low secondary to suppression of renin by excess DOC, as in the case of 11β-hydroxylase deficiency. The 17α-hydroxylase-deficient patients do not experience adrenal crisis despite inadequate cortisol synthesis. Overproduction of corticosterone provides adequate physiologic response to stress. Plasma ACTH levels are less elevated than in other conditions of impaired cortisol production. Gonadotropin production is extremely elevated in both sexes because of the absence of any sex steroid feedback; the gonads are atrophic.
(i.e., compound B) and the metabolites of these two steroids.14 Aldosterone is often low secondary to suppression of renin by excess DOC, as in the case of 11β-hydroxylase deficiency. The 17α-hydroxylase-deficient patients do not experience adrenal crisis despite inadequate cortisol synthesis. Overproduction of corticosterone provides adequate physiologic response to stress. Plasma ACTH levels are less elevated than in other conditions of impaired cortisol production. Gonadotropin production is extremely elevated in both sexes because of the absence of any sex steroid feedback; the gonads are atrophic.
A high ratio of Δ5 to Δ4 steroids characterizes the 3β-HSD deficiency.29 Serum levels of 17-hydroxypregnenolone and DHEA are elevated before and after ACTH stimulation. Increased excretion of the Δ5 metabolites pregnanetriol and 16-pregnanetriol in the urine is also diagnostic for this enzyme disorder.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


