Chapter 26 Darunavir
INTRODUCTION
Protease inhibitors (PIs) have played a crucial role in the history of antiretroviral therapy. Their use as components of highly active antiretroviral therapy (HAART) has been shown to result in durable virologic suppression and dramatic improvement in the morbidity and mortality of patients infected with the human immunodeficiency virus type 1 (HIV-1).1–3 Thus, PIs have become cornerstones of drug treatment for HIV, whether as first-line agents for treatment-naive patients or as alternative agents for patients with a long history of antiretroviral therapy, with previous virologic failures and resistance mutations.
The emergence of drug-resistant strains of HIV-1 represents a significant threat to the efficacy of antiretroviral therapy, with increasing levels of resistance4 and the extensive cross-resistance that exists between currently available PIs limiting the sequential use of these agents. The rational development of novel therapies, which effectively suppress viremia and address the complex problems of resistance, offers a way forward for HIV therapy.5–7 The novel PI, darunavir (DRV; also known as TMC114 (Prezista)), was designed to be active against both wild-type HIV and strains that are resistant to currently available PIs. A lead optimization program produced a series of compounds, from which DRV was selected for development because of its favorable pharmacokinetic (PK) and antiviral profile.6,7 As for many other PIs, the PK profile and clinical effectiveness of DRV were found to be improved by PK enhancement with low-dose ritonavir (RTV).8
CHEMICAL STRUCTURE
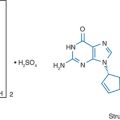
DRV is a novel, potent, HIV-1 PI that contains a 3 (R),3a (S),6a (R)-bis-tetrahydrofuranylurethane (bis-THF) isostere.9 The method of synthesis of DRV was first published by Ghosh and colleagues in 2004.10 Figure 26-1 shows the chemical structure of DRV.

Figure 26-1 Chemical structure of DRV.
Adapted from King NM, Prabu-Jeyabalan M, Nalivaika EA, et al. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol 78:12012–12021, 2004.
Although DRV shares some structural similarities with amprenavir (APV), it binds ∼100 times more tightly than APV to the HIV-1 protease (Fig. 26-2). This is due in part to the additional hydrogen bonds formed between the bis-THF moiety in the P2-pocket of DRV and the protease molecule.5,7 Furthermore, DRV binds particularly tightly due to its high affinity with main-chain atoms at the bottom of the active site.5 It has been shown that, unlike currently available PIs, DRV shows a fast association but very slow dissociation from wild-type HIV-1 protease.11 Another characteristic of DRV is that it binds predominantly within the substrate consensus volume (the region of the enzyme’s active site occupied by substrates) of HIV-1 protease. The structural shape of this volume is critical to HIV protease activity and any mutation that affects the fit of DRV would likely render the enzyme inactive, thereby preventing further viral replication. Thus, the close conformation of DRV within the substrate consensus volume may be an important factor in its low susceptibility to viral resistance.5
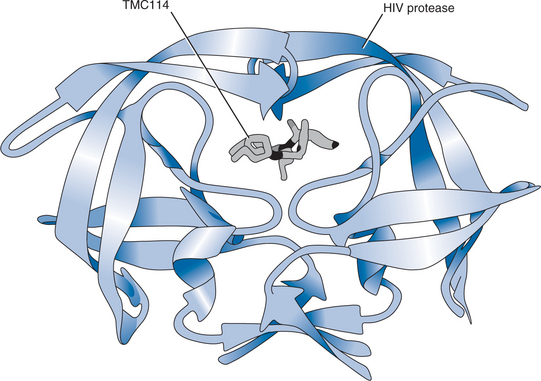
Figure 26-2 DRV in the binding pocket of the HIV-1 protease dimmer, shown as ribbon diagrams.
Adapted from King NM, Prabu-Jeyabalan M, Nalivaika EA, et al. Structural and thermodynamic basis for the binding of TMC114, a next-generation human immunodeficiency virus type 1 protease inhibitor. J Virol 78:12012–12021, 2004.
High-resolution crystallographic analysis has been performed on DRV in complexes with wild-type HIV-1 protease and the mutant proteases PRV82A and PRI84V that are common in drug-resistant HIV.12 DRV forms hydrogen bonds with the conserved main-chain atoms of Asp29 and Asp30 of the protease. The close contact of DRV with the main chains of these active-site amino acids is not seen with other currently available PIs and it is believed to be critical for the potency of DRV against multiple PI-resistant HIV isolates.9
Furthermore, the structural flexibility of DRV indicates that small changes in the substrate consensus volume may not impair the overall ability of the drug to bind tightly to the enzyme, thus, conferring additional resilience to the effect of PI resistance-associated mutations.5
IN VITRO ACTIVITY
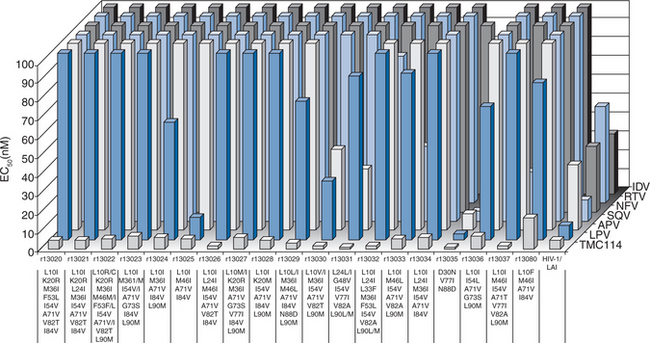
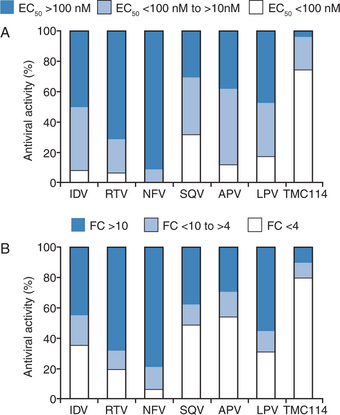
A lead optimization program resulted in DRV being identified and selected for development based on favorable PK characteristics in animals and potent in vitro activity against a comprehensive panel of clinical isolates of HIV representative of those found in clinical practice. De Meyer and colleagues characterized the antiviral activity of DRV against both wild-type and PI-resistant HIV.13 Against wild-type HIV, DRV had a 50% effective concentration (EC50) of 1–5 nM and an EC90 of 2.7–13 nM. DRV exhibited no cytotoxicity at concentrations up to 100 μM, resulting in a selectivity index of >20 000 for wild-type HIV. DRV were also active against all viruses in a panel of 19 recombinant clinical isolates that were assembled to evaluate newly synthesized compounds (the viruses carried multiple protease mutations and were resistant to an average of five other PIs with resistance defined as a fold change in EC50 (FC) ≥4). Figure 26-3 shows the activity of DRV and six currently approved PIs against PI-resistant and wild-type HIV-1.13 DRV and six approved PIs were also tested against a broad selection of several thousand HIV clinical isolates exhibiting various degrees of resistance to all commercially available PIs. DRV had significantly greater potency against the 1501 samples with PI resistance than any other tested PI (Fig. 26-4). Antiviral analyses performed on DRV by Koh and colleagues yielded similar findings to those described above.9
HIV-1 group M subtype B is currently predominant in the USA and Europe. However, studies have suggested that the prevalence of non-B subtypes and circulating recombinant forms may soon reach significant levels.14,15 DRV showed similar potent activity against a range of clinically derived recombinant viruses representing HIV-1 group M subtypes A through H, and including a number of recombinant forms of HIV-1 group O.13
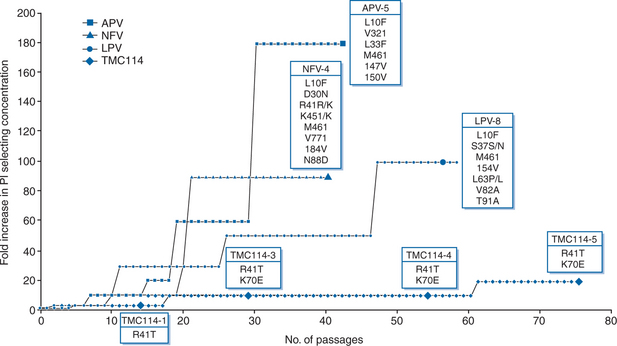
The results of in vitro selection experiments showed that the emergence of resistant HIV was much slower with DRV than with comparator PIs APV, nelfinavir, or lopinavir (LPV) (Fig. 26-5).13 This indicates an increased genetic barrier to the development of resistance to DRV and a pathway to resistance that is different from that of other currently available PIs.
PHARMACOKINETICS
Effect of Co-Administration with Low-Dose Ritonavir
A comparison of two randomized, multiple dose-escalating phase I trials in a total of 76 healthy volunteers concluded that co-administration of DRV and low-dose RTV (DRV/r) resulted in improved PK characteristics compared with DRV alone despite the use of lower doses of DRV.8 Co-administration of RTV also resulted in improved safety and tolerability profiles. Both trials used an early experimental formulation of DRV as a polyethylene glycol (PEG) 400-containing oral solution. In the first trial, unboosted DRV was administered (in a placebo-controlled, double-blind manner) at doses of 400 mg twice daily, 800 mg twice daily, 800 mg three times daily and 1200 mg three times daily, while in the second (open-label) study, DRV/r was administered at doses of 200/100 mg once daily, 400/100 mg once daily, 300/100 mg twice daily, 600/200 mg once daily and 1200/200 mg once daily. DRV was rapidly absorbed with a time to maximum plasma concentration (tmax) achieved in less than 3 h and steady-state concentrations were reached within 3 days. Overall, mean maximum plasma concentration (Cmax), minimum plasma concentration (Cmin) and average steady-state plasma concentration (Css,av) achieved at day 14 were higher with DRV/r than with DRV alone. With DRV alone, mean Cmin at day 14 ranged from 14 ng/mL for the lowest dose to 142 ng/mL for the highest dose. The corresponding ranges for Cmax and Css,av at day 14 were 2168–8040 ng/mL and 270–1395 ng/mL, respectively. In the RTV-boosted trial, values for Cmin ranged from 480 to 1486 ng/mL, Cmax from 1569 to 5453 ng/mL and Css,av from 725 to 2460 ng/mL. The PK results demonstrated that the values for Cmin and the area under the plasma concentration-time curve (AUC) at 24 h for DRV are substantially increased with the co-administration of low doses of RTV. Higher Cmin concentrations of DRV were obtained at lower total daily doses of DRV with addition of RTV.
Effect of Food
In early studies, DRV was administered as an oral solution. However, a solid tablet formulation was developed for use in phase II dose-finding trials of DRV/r. A PK study confirmed that under fed conditions and with low-dose RTV, the tablet and oral solution of DRV meet the criteria for bioequivalence.16 The study also showed that in the presence of low-dose RTV, the bioavailability of DRV oral tablets is greater after feeding than after fasting. It is therefore recommended that DRV tablets should be taken with food. Another study confirmed that in the presence of low-dose RTV, systemic exposure to DRV is increased by ∼30% when tablets are taken after a meal than after fasting.17 PK parameters in the study were determined after administration of a single oral dose of 400 mg of DRV in tablet formulation together with low-dose RTV under fasted conditions and with different types of food (standard breakfast, high-fat breakfast, protein-rich nutritional drink and croissant with coffee). The results demonstrated that the type of meal consumed does not affect the exposure to DRV.
Effect of Increased Intragastric pH
The effects of concomitant administration of DRV/r with drugs that inhibit gastric acid secretion and thereby elevate intragastric pH have been investigated. The combination of the proton-pump inhibitor omeprazole (20 mg daily) or the H2-receptor antagonist ranitidine (150 mg twice daily) with DRV/r did not affect the PK characteristics of DRV after multiple dosing.18
EFFICACY
The efficacy of DRV/r has been studied in clinical trials in treatment-experienced, HIV-infected patients. An overview of the design and key features of trials providing clinical efficacy data is shown in Table 26-1.






Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


