Chapter 50 Cytomegalovirus Disease
Cytomegalovirus (CMV) is a common infection in the general community, with 60% of adults in developed countries typically seropositive, rising to 90–100% in developing countries and in poorer socioeconomic groups within developed countries. CMV is highly immunogenic and a considerable proportion of the human immune response is directed to keeping this virus suppressed into latency. It follows that end-organ disease due to CMV occurs late in the course of human immunodeficiency virus (HIV) infection, once HIV has progressively damaged immune capability. The increased use of highly active antiretroviral therapy (HAART) for the treatment of HIV infection has led to improved immunocompetence and a decrease in the incidence of opportunistic infections, including CMV, in persons with HIV infection.1 Immune reconstitution due to HAART is the most important therapeutic intervention for prevention and for long-term management of established CMV disease in persons with HIV infection. However, CMV-associated diseases are still seen for a variety of reasons (see Table 50-1).
Table 50-1 Some Reasons Patients Continue to Present with CMV End-Organ Disease in the Era of HAART
| Not Taking HAART |
| Unaware of HIV infection Lack of access to healthcare Unwilling to take medication |
| Failure of HAART |
| HIV resistance Poor compliance |
PATHOGEN
CMV was first isolated in 1956 using the ‘new technology’ of cell culture in vitro. The only cells to support growth were human fibroblasts and the evolution of cytopathic effect in these cells was slow. This slow growth became part of the definition of the Betaherpesvirinae subfamily of Herpesviridae of which CMV is the prototype member. This slow growth in vitro, coupled with the absence of overt disease in people with normal immunity and restriction of disease to those with impaired immunity such as the fetus or allograft recipient, led to the impression that CMV is a virus of low inherent pathogenicity. The application of more modern technology in the form of quantitative polymerase chain reaction (PCR) to measure changes in viral load directly in humans2 has shown that this impression is false and is an artifact of propagating the virus in fibroblasts in vitro.
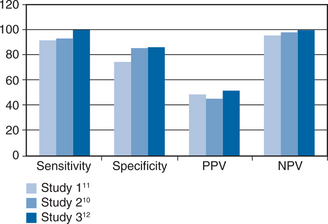
Studies of CMV titer measured in the urine of children with congenital CMV first reported in 1975 that CMV disease was associated with a high viral load.3 The implication from this report, that CMV disease developed once a threshold value of viral load had been crossed, was confirmed in 1997 in renal allograft patients when multivariate statistical analyses showed that a high viral load explained the previously recognized risk factors of viremia (detected by cell culture) and seropositive donor serostatus.4 The same was true when CMV viral load was measured in the blood of renal transplant recipients and the measurement of such viremia (viremia throughout this chapter refers to virus detected in the blood by PCR) completely replaced detection of CMV in urine because viremia has a closer correlation with disease events.5–7 Viremia was also strongly associated with CMV disease in liver transplant patients and stem cell transplant patients where it again explained the previously recognized risk factors.8,9 These principles of CMV pathogenesis in the immunocompromised host transferred directly to acquired immunodeficiency syndrome (AIDS) patients when, in 1997, three groups10–12 simultaneously reported that viremia preceded CMV retinitis (see Fig. 50-1). However, there is one differentiating feature between these groups of immunocompromised hosts; in AIDS patients, CMV crosses the blood–organ barrier to preferentially cause clinically relevant retinal disease whereas this is uncommon in allograft recipients. Specifically, 85% of AIDS patients with CMV end-organ disease have retinitis (with ∼10% having enteritis and the remaining patients having neurological disease); whereas, retinitis occurs in less than 5% of allograft recipients.13,14 This striking difference between these groups of patients remains unexplained. Nevertheless, it is important to consider the concept of viremic dissemination of CMV in AIDS patients with retinitis because viremia is the route by which the virus reaches the contralateral eye and other organs.
Direct measurements have been made of the turnover of CMV in serial samples of viremic patients and reveal that CMV replicates with rapid dynamics.2 Specifically, in patients with primary CMV infection, the dynamics are essentially equivalent to those of primary HIV infection.15 One important difference from HIV replication however, is that CMV DNA polymerase has a proofreading activity so that many replicative cycles are required to select for resistance to drugs such as ganciclovir (GCV). Strains resistant to GCV frequently coexist with wild type strains in clinical samples. These strains compete in the laboratory so that, in the absence of ganciclovir, the wild type out-competes the mutant, leading to failure to identify resistant subpopulations.16
IMMUNOLOGY
Cytomegalovirus interferes with this pathway at several points (see Fig. 50-2). First, a phosphoprotein termed pp65 phosphorylates the major immediate early antigen of CMV so that this cannot be degraded in the proteosome.17 This may explain why pp65 is the immunodominant protein of CMV seen by the cell-mediated immune system, rather than the major immediate early protein which is the first protein to be made in the cell. This might represent a mechanism of the virus to divert immune responses away from important proteins and toward others that are less able to control CMV infection. Protein US6 blocks TAP, protein US3 binds to HLA class I molecules and sequesters them in the ER, protein US2 and protein US11 mimic the natural cellular pathway that reexports class I molecules into the cytosol leading to their degradation.18–22 These proteins act in a coordinated temporal fashion, with pp65 being a structural protein of the virus released by uncoating, so that it is active as soon as the virus enters the cell, US3 being active at immediate early times and US6, US2, and US11 being active at early times. Thus, before the onset of DNA replication, CMV has taken over the cell and effectively prevented its recognition by the CTL. However, the cell is still susceptible to attack from NK cells and macrophages which sense the reduced display of HLA class I (missing self hypothesis). CMV has evolved genes to combat these responses as well. Gene UL18 contains an HLA class I homolog (presumably captured from the human genome millions of years ago) which is presented at the plasma membrane and provides an inhibitory signal to macrophages through the CD85j receptor.23,24 Another HLA class I homolog is encoded by gene UL142 which provides a negative signal to NK cells through an unknown molecule.25 Gene UL40 contains a 9 amino acid peptide which is identical to the signal peptide of HLA class I molecules. This is presented by HLA-E, so mimicking the natural authentic signal which NK cells sense through the CD94 receptor, and provides an inhibitory signal by upregulating HLA-E.26,27
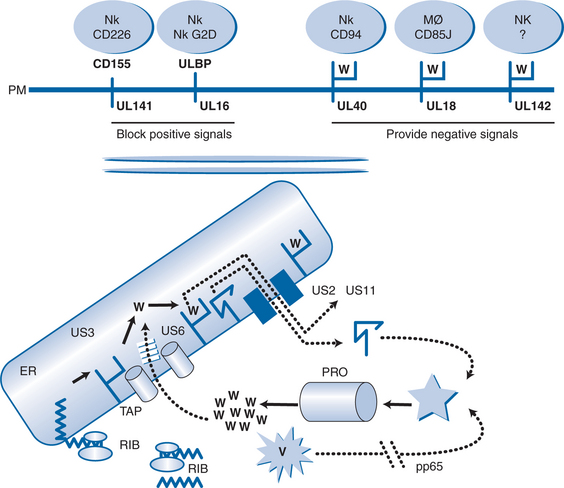
In addition to these negative signals, CMV interferes with natural positive signals given to NK cells via their G2D receptor. Gene UL16 is expressed and binds to natural gene products (dubbed UL binding proteins) as well as MICA and MICB which normally provide a positive signal to the NK cell through the G2D receptor.28 The protein from gene UL141 binds CD155, so preventing it from giving an activating signal through CD226.26
The net result of all of these immune evasion genes is that CMV can replicate in cells relatively free from attack from the CTL, NK, and macrophage components of the immune system. The induction phase of the immune system is fully active and a considerable proportion of the host immune response is directed toward CMV-encoded epitopes. For example, using tetramer reagents, typically 1% of CTLs recognize a single peptide from pp65 protein.29 In normal individuals, a stand-off occurs between this high incidence of CTLs and the sequestered nature of CMV replicating in relatively protected cells. However, when HIV progressively damages the effectiveness of CTLs, CMV gains the upper hand and can replicate to high levels causing viremia and subsequent dissemination in the bloodstream.
Several authors have described CMV-specific cell-mediatedimmune responses which correlate with protection against CMV viremia30 or delayed progression of retinitis.31 These cells include a broad range of antigenic diversity repertoire and degree of differentiation;31 immediate-early specific, interferon gamma (IFNγ) producing CD8+ T-lymphocyte cells,32,32 IFNγ CD4+ T-lymphocyte cells33 or ELISpot positive results, but not IFNγ production30 were each reported to be associated with a better prognosis. Furthermore, some others32 reported maintenance of large populations of CMV-specificCD81 T-lymphocyte cells in the absence of specificCD41 T-lymphocyte cells. Thus, it is not possible to identify at this time, the immune correlates of protection against CMV infection and disease.
EPIDEMIOLOGY OF CMV END-ORGAN DISEASE
In HIV-infected persons, as with most other opportunistic infections associated with HIV infection, CMV disease appears in a previously infected host. This may be due to reactivation of the latent virus although reinfection with a different strain is an additional mechanism. Although more than 90% of persons with HIV infection have antibodies to CMV, indicating prior infection, the clinical manifestations of CMV disease do not generally present until the CD4+ T-lymphocyte count drops below 100 cells/μL.34–36 In a multicenter observational cohort of 1002 HIV-infected persons with fewer than 250 CD4+ T lymphocytes per microliter receiving zidovudine, disease due to CMV developed in 109 persons.14 Kaplan–Meier estimates of the proportion of persons who developed CMV disease was 21.4% at 2 years for persons entering the study with fewer than 100 CD4+ T lymphocytes per microliter and 10.3% for persons with initial counts higher than 100 cells/μL. A smaller study of 135 persons with fewer than 250 CD4+ T lymphocytes per microliter found the Kaplan–Meier estimate for the development of CMV retinitis to be 42% within 27 months for the group with fewer than 50 CD41T lymphocytes per microliter.37 Of the 26 persons developing CMV retinitis, 24 had CD1 T-lymphocyte counts of less than 50 cells/μL before developing CMV retinitis; the other two persons had counts of 60 and 160 cells/μL 7 and 11 months before the diagnosis, respectively. The mean time from the first CD4+ T-lymphocyte count of less than 50 cells/μLuntil the diagnosis of CMV retinitis was 13.1 months. Since 1990, when cases of Pneumocystis carinii (now termed Pneumocystis jiroveci) pneumonia (PCP) reported to the CDC began to decrease owing to the widespread use of prophylaxis to prevent PCP, cases of AIDS-defining CMV retinitis increased. (Total cases of CMV retinitis, however, did not decrease.) A prospective cohort of 844 men were followed before the development of an AIDS-related opportunistic infection. Among those who received PCP prophylaxis, CMV disease was the initial AIDS-related opportunistic infection in 9.4% of men compared to 3.1% of men who did not receive PCP prophylaxis. The lifetime occurrences of CMV disease were 44.9% and 24.8% in those groups, respectively.38
Disease due to primary infection with CMV is rarely recognized in persons with HIV infection. More than 90% of persons with AIDS in the United States have evidence of prior infection with CMV.39 Reactivation of the latent infection may result in systemic signs and symptoms such as fevers, myalgias, leukopenia, and weight loss in addition to symptoms attributable to end-organ disease.
GENERAL LABORATORY DIAGNOSIS
Data on the use of a qualitative CMV-specific polymerase chain reaction (PCR) on whole blood suggests that this test may be both sensitive and specific for the development of CMV disease. In a prospective cohort of 97 HIV-infected persons with fewer than 50 CD4+ T lymphocytes per microliter followed every 3 months with CMV PCR, 16 of 27 persons (59%) who were CMV-positive by PCR at baseline developed CMV disease within 12 months compared with only three of 70 persons (4%) who were initially CMV negative.12 In a cohort of 94 HIV-infected individuals without CMV disease at baseline, a qualitative plasma CMV PCR was more sensitive and specific (89% and 75%, respectively) than either urine CMV cultures (85% and 29%, respectively) or leukocyte cultures (38% and 74%, respectively) for the identification of persons developing CMV disease within 12 months.11 Quantitative CMV PCR was able to increase the specificity of the assay at some cost to the sensitivity.
In an analysis of 619 persons with AIDS who participated in a study of the efficacy of oral GCV for the prevention of CMV disease, CMV DNA was quantitated by PCR.40 A positive baseline plasma CMV DNA PCR was associated with a 2.5-fold risk of death, and each log increase in CMV DNA load was associated with a 3.1-fold increased risk for CMV disease and a 2.2-fold increase in mortality (p < 0.001 for both).40 Commercial PCR assays for CMV are currently available, but these study data have not translated into widespread clinical use in HIV-infected persons. The identification of persons at high risk for the development of CMV disease may allow the development of preemptive therapeutic strategies to prevent end-organ disease due to CMV; but because of the overwhelming impact of HAART in the management of persons with advanced HIV infection, such strategies are of marginal use at present. A prospective study (ACTG 5030) is assessing the utility of plasma PCR for preemptive therapy and its results are awaited with interest.
NATURAL HISTORY AND ORGAN-SPECIFIC DIAGNOSIS
Retinitis
Retinitis due to CMV results from the hematogenous dissemination of CMV after reactivation of a latent CMV infection.34 Progression of the infection within the retina is generally to contiguous cells. Lesions near the macula or optic nerve (zone 1) commonly produce complaints of decreased visual acuity or defects in the visual field. Retinal lesions at least 1500 μm from the edge of the optic nerve and at least 3000 μm from the center of the fovea (zones 2 and 3) or anterior to the equator of the eye may be asymptomatic or present with the complaint of ‘floaters’ or loss of peripheral vision. CMV retinitis is not associated with pain or photophobia.
Visual loss due to CMV retinitis occurs in several ways. Direct infection of the retinal cells by CMV causes retinal necrosis, which may result in a visual field defect or scotoma, depending on where in the retina the lesion occurs. This permanent, irreversible loss of vision is not amenable to therapy and depends on the location and extent of the retinal necrosis. Normal central vision may be preserved if the macula is not involved. Second, retinal involvement of the area near the macula may produce edema in the macula and loss of central visual acuity. The macular edema and loss of visual acuity are potentially reversible if recognized and treated promptly before the retinal cells are infected. Third, after infection with CMV and subsequent retinal necrosis, the retina is left as a thin, atrophic tissue that is susceptible to breaks and detachment.41 Retinal detachment occurs commonly in persons with CMV retinitis and presents with the sudden onset of floaters, flashing lights, loss of visual field, and decreased visual acuity. Retinal detachments can be repaired; but owing to the nature of the atrophic retinal tissue they frequently recur. Data from 681 patients with CMV retinitis reported substantial visual loss on follow-up. Events per 100 eye-years ranged from 51.7 to 97.7 for loss of visual acuity to worse than 20/40 and 18.9 to 49.1 for acuity worse than 20/200.42
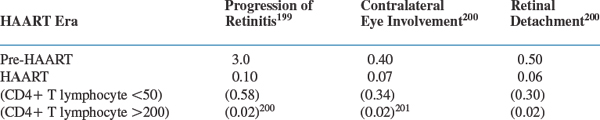
Recent data show that the incidence of progression of retinitis, retinal detachments, and involvement of the contralateral eye is markedly reduced in persons receiving HAART (see Table 50-2). Nevertheless, CMV retinitis, whether acute or long-standing, continues to significantly affect quality of life when measured formally.43
Table 50-2 Clinical Event Rates (Per Person-Year of Follow-Up) Reported from a Prospective Cohort of 271 Patients with CMV Retinitis
Data from randomized controlled trials using standard treatment with GCV, foscarnet, or cidofovir showed that the median time to relapse after successful initial therapy of CMV retinitis, in the absence of HAART, ranges from 50 to 120 days.44–46
Because of the difficulty of obtaining retinal tissue for histopathologic examination, CMV retinitis is diagnosed based on the appearance of the characteristic perivascular fluffy yellow-white retinal infiltrate that is often associated with retinal hemorrhage. The portion of the vessels near the lesion may appear to be sheathed. Occasionally, the lesions present with a granular, rather than fluffy, appearance.34 Progression of retinitis is in a characteristic ‘brushfire’ pattern, with a granular, white leading edge advancing before an atrophic, gliotic scar. Progression is irregular and occurs in fits and starts. In a study performed before the availability of HAART using serial, masked retinal photographs, the median progression rate at which disease approached the fovea in 17 untreated patients was found to be 24.0 μm/day compared with a median progression rate of 11.5 μm/day in 14 patients treated with GCV.47 Patients with AIDS and CMV retinitis, who are not being treated with HAART and have not reconstituted their immune system, usually have minimal inflammation of the vitreous. CMV retinitis usually presents unilaterally but, untreated, becomes bilateral in most cases. Retinitis in a CMV-seronegative individual would make a diagnosis of CMV retinitis highly improbable.
Other ocular lesions that occur in HIV-infected persons are in the differential diagnosis of CMV retinitis.48 Cotton-wool spots are microinfarctions of the retinal nerve fiber layer that occur commonly in persons with HIV infection and may present similar to an early CMV retinitis.49 These small, white lesions do not affect vision and spontaneously regress over several weeks. In the setting of an HIV-infected person with fewer than 100 CD4+ T lymphocytes per microliter, it is critical to ensure that lesions that appear to be cotton-wool spots regress and do not progress as would be expected with CMV retinitis. Acute retinal necrosis, caused by herpes zoster, presents with either peripheral retinal vascular occlusion overlying a white, necrotic retina or, when more central vessels are involved, ischemia due to central vascular occlusion.50 Intraocular lymphomas may present with small retinochoroidal infiltrates, optic nerve head swelling, and vascular sheathing. Toxoplasmic chorioretinitis may be unifocal or multifocal but is usually associated with a moderate to severe inflammatory reaction in the vitreous, which helps differentiate it from CMV retinitis.51 Pneumocystis jiroveci chorioretinitis appears as multifocal, white-yellow raised choroidal lesions with minimal inflammation.52 Other, rare causes of ocular disease in persons with HIV infection include syphilis, Mycobacterium tuberculosis, Cryptococcus, and Candida infections, and histoplasmosis.53
Gastrointestinal Disease
CMV Esophagitis
Esophagitis due to CMV is a common cause of odynophagia in persons with AIDS.53 Candida more characteristically causes dysplasia. CMV esophagitis occurs in ∼10% of persons with AIDS who develop CMV disease,14 but is much less common than Candida esophagitis. The definitive diagnosis of CMV esophagitis is established by biopsy evidence of CMV with an inflammatory response in the appropriate clinical setting. The presence of extensive large, shallow ulcers of the distal esophagus is the hallmark of the disease. Pathologically, the large intranuclear inclusion bodies characteristic of CMV can be seen in the endothelial cells at the edge of the ulcer and are required to confirm the diagnosis.54 Immunohistochemical stains may add to the sensitivity of routine hematoxylin and eosin staining for CMV. Culturing CMV from a biopsy or brushing of the esophagus is not sufficient to establish the diagnosis of CMV esophagitis because many persons with low CD4+ T-lymphocyte counts are viremic and have positive cultures for CMV in the absence of clinical disease.55 Furthermore, adding viral culture and brush cytology to routine multiple biopsies with standard histopathology does not increase diagnostic sensitivity.56
The differential diagnosis of CMV esophagitis includes esophagitis due to herpes simplex, reflux or peptic ulcer disease, histoplasmosis, Kaposi sarcoma, lymphoma, HIV infection, M. tuberculosis infection, and rarely Mycobacterium avium-intracellulare complex infection, cryptosporidial infection, and Pneumocystis jiroveci pneumonia.57 Because of the relative prevalence of Candida esophagitis in this population, many clinicians treat esophageal symptoms empirically, especially in the presence of oral thrush. Empiric therapy usually consists of fluconazole for presumptive Candida esophagitis, endoscoping only those persons who fail to respond.58 Both CMV or Candida esophagitis most commonly present with CD4+ T-lymphocyte counts of less than 50 cells/μL; however, persons with esophageal candidiasis, but not CMV, can present with counts between 50 and 400 cells/μL.36,45
CMV Gastroduodenitis
Cytomegalovirus may be an under-recognized cause of peptic ulcers in AIDS patients, in whom it is found more frequently than in association with Heliobacter pylori.59
CMV Colitis
Colitis due to CMV occurs in fewer than 10% of persons with AIDS in whom disease due to CMV is diagnosed.14 Fever, weight loss, anorexia, abdominal pain, debilitating diarrhea, and malaise are frequently present. Extensive hemorrhage and perforation can be life-threatening complications.60 The symptoms are nonspecific and may be similar to those due to other gastrointestinal pathogens, such as Cryptosporidium, Microsporidium, Cyclospora cayetanensis, Mycobacterium avium complex, Giardia lamblia, Entamoeba histolytica, Salmonella, and Shigella or the gastrointestinal involvement seen with lymphoma or Kaposi sarcoma. The radiographic manifestations of CMV colitis are nonspecific and may mimic the findings of other inflammatory bowel conditions, including ulcerative colitis.61 Colonoscopic or rectal biopsy with histopathologic identification of characteristic intranuclear inclusions is required for diagnosis. Identification of CMV by culture or even on histopathologic specimens may not be sufficient to implicate CMV; frequently multiple pathogens may coexist and must be considered in persons with advanced HIV infection.62
Pneumonitis
Although CMV can be cultured routinely from throat washings, pulmonary secretions, bronchoalveolar lavage specimens, and autopsy lung tissue, CMV is seldom implicated antemortem as an isolated pathogen causing pneumonitis in persons with HIV infection.63 Foci of CMV inclusion bodies and pneumonitis are often found at autopsy. In one study from San Francisco, although CMV was cultured from bronchial alveolar lavage fluid or transbronchial biopsy specimens in 54 of 111 patients diagnosed with their first episode of PCP, the presence of CMV had no impact on the long-term survival, acute death rate, or length of hospital stay.64 Of 17 persons with biopsy-diagnosed CMV pneumonitis, no clinical, radiographic, or histologic findings distinguished persons with CMV as the sole pathogen from those with other, concomitant pathogens.65
Neurologic Disease
Cytomegalovirus has been found to be associated with various neurologic infections in persons with HIV infection, particularly ventriculoencephalitis66–72 and ascending polyradiculopathy.73–76 Ventriculoencephalitis usually occurs in advanced HIV infection in persons with a prior CMV disease diagnosis. Patients typically present with lethargy, confusion, and fever; but the clinical presentation may overlap that of HIV encephalitis. The cerebrospinal fluid (CSF) generally shows a pleocytosis that may be polymorphonuclear, low to normal glucose levels, and normal to elevated protein levels. PCR techniques are superior to culture for detection of CMV in CSF.67,72 Periventricular enhancement of computed tomography (CT) or magnetic resource imaging (MRI) scales are suggestive of CMV ventriculoencephalitis rather than HIV-related neurologic disease.
Polyradiculopathy caused by CMV is characterized by urinary retention and progressive bilateral leg weakness.73–76 The clinical symptoms generally progress over several weeks to include loss of bowel and bladder control and flaccid paraplegia. A spastic myelopathy has been reported, and sacral paresthesia may occur. The CSF often shows a pleocytosis that is usually polymorphonuclear, hypoglycorrhachia, and elevated protein levels. PCR techniques are superior to culture for detection of CMV in CSF.
Although CMV is found in the central nervous system (CNS) in up to 25% of persons in autopsy studies,66,71,77 the incidence of overt CMV neurologic disease antemortem appears to be low.
Other CMV-Associated Diseases
CMV Adrenalitis
Involvement of the adrenal glands with CMV is frequently reported in autopsy studies of persons with HIV infection, with the involvement documented in as many as 64 of 83 (77%)78 and 42 of 71 (59%)77 persons in two autopsy studies. Patients rarely manifest adrenal insufficiency by laboratory or clinical parameters. Adrenalitis due to CMV is uncommonly diagnosed premortem, but hypoadrenalism is occasionally documented in persons with CMV disease by the cosyntropin stimulation test.79
CMV Hepatitis and Biliary Disease
Although CMV involvement of the liver and biliary tract is often seen in autopsy specimens, clinical hepatitis due to CMV is rare in persons with HIV infection.14 Biliary tract or hepatic involvement by CMV may present with right upper quadrant pain and elevated alkaline phosphatase, but infections with Cryptosporidium and M. avium-intracellulare complex are more common causes. In one series of 66 consecutive persons with AIDS and first-episode gastrointestinal CMV disease, 22 patients presented with esophagitis, 28 with colitis, nine with sclerosing cholangitis, and only two with acute hepatitis.80
THERAPY
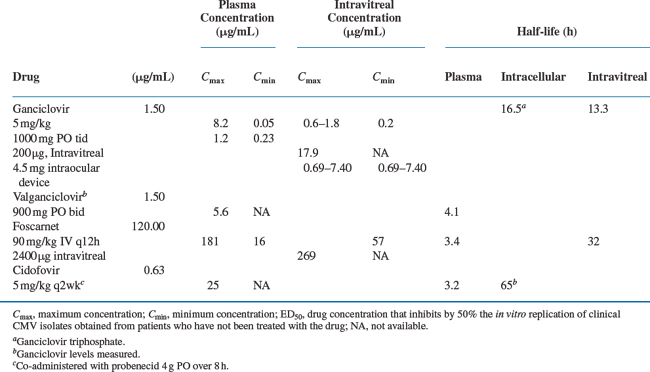
Specific therapies for CMV disease are summarized inTable 50-3. Drug susceptibilities and pharmacokinetic values are summarized in Table 50-4. The chemical formulas of the drugs are shown in Figure 50-3.
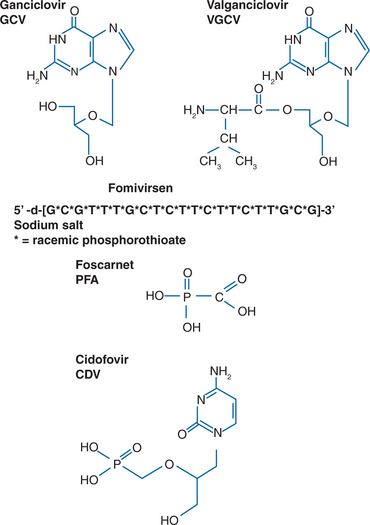
Specific Treatments
The US Food and Drug Administration (FDA) has approved seven approaches to CMV therapy for patients with HIV (Table 50-5). Oral GCV is no longer available having been replaced by valganciclovir (VGCV) oral prodrug, and we will give the equivalent dose of VGCV where appropriate.
| Drug | Year of FDA Approval |
|---|---|
| Ganciclovir IV | 1988 |
| Foscarnet IV | 1991 |
| Ganciclovir capsules | 1994a |
| Ganciclovir implant | 1996 |
| Cidofovir IV | 1996 |
| Fomivirsen injectable | 1998 |
| Valganciclovir | 2001 |
Ganciclovir
Initial Therapy with Intravenous GCV
The first major advance in the treatment for CMV disease was the development of GCV, an agent highly specific for human herpesviruses. GCV is a nucleoside analog whose activity depends on inhibition of herpesvirus DNA polymerases. It requires phosphorylation in CMV-infected cells (see Fig. 50-4), and most strains of CMV resistant to GCV are unable to phosphorylate GCV. The CMV UL97 open reading frame codes for a protein kinase capable of phosphorylating GCV in CMV-infected cells.81,82 When treatment for disease is stopped, viral spread and progression of disease characteristically begin again.35 Prior to the introduction of HAART, lifelong daily therapy was required, most often intravenously through an indwelling catheter. An oral formulation of GCV was used, but was poorly absorbed (less than 10% bioavailable) and not as clinically effective as the intravenous formulation.83,84
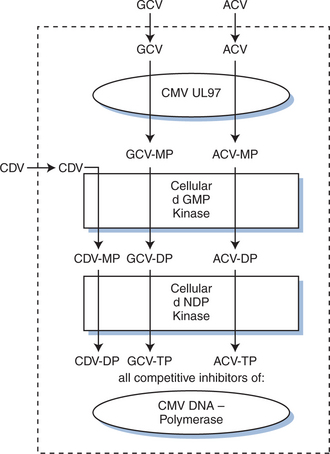
When given intravenously by a 1-h infusion, the standard 5 mg/kg dose of GCV reaches a maximum concentration in the plasma at the end of infusion of ∼6 μg/mL (24 μM).84 Trough levels 11 h after infusion are ∼1 μg/mL (4 μM). The initial distribution half-life (t½) is ∼0.76 h, and the terminal elimination t½ is 3.60 h.84 Most studies report that for human CMV isolates the 50% inhibition (ID50) of viral plaque formation is attained by concentrations of GCV between 0.4 and 11.0 μM.35,85
GCV was licensed by the FDA in 1988. The recommended dosing of GCV for the treatment of CMV retinitis in persons with AIDS is 5 mg/kg IV twice daily for a 14- to 21-day induction period followed by a 5 mg/kg daily indefinite maintenance phase. The terms ‘induction’ and ‘maintenance’ may be misnomers inasmuch as progression of CMV retinitis is regularly seen during the maintenance phase with GCV and foscarnet (Table 50-3). Patients with progression during the maintenance phase are routinely retreated with the twice-daily regimen. GCV at 1000 mg PO thrice daily was approved for maintenance therapy of CMV retinitis but was not recommended for persons whose central visual acuity would be threatened if progression of disease should occur. VGCV was licensed by the FDA in 2001 and is the oral anti-CMV agent of choice.
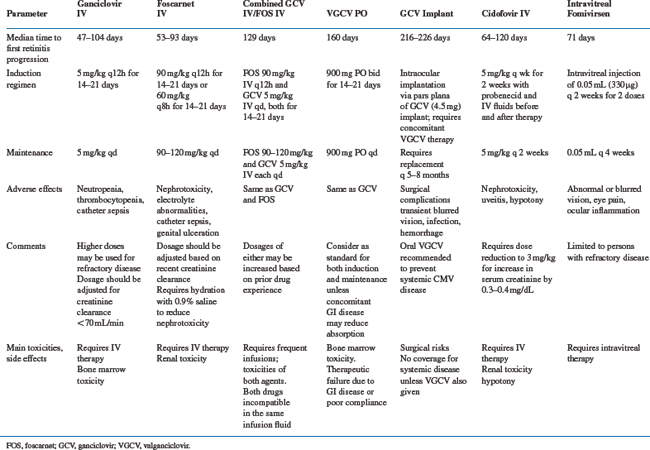
In a compilation of clinical data, treatment with GCV resulted in the improvement or stabilization of CMV retinitis in 80–90% of patients.86 The median time to clinical progression in these uncontrolled studies appeared to be as long as 145 days from the diagnosis of CMV retinitis while continuing some maintenance therapy with GCV. One large, uncontrolled series reported the outcomes of 105 immunocompromised (primarily AIDS) patients who were treated with GCV for CMV retinitis. Analysis of a subset of these patients selected for their ability to tolerate a prolonged course of therapy and who had high-dose maintenance therapy (25–35 mg kg−1 week−1) after induction therapy showed that the mean time to progression of retinitis was 18 weeks. This result, however, is based on subset analysis; these patients were selected for their ability to tolerate a prolonged course of relatively high-dose GCV without the development of neutropenia. The results of other trials indicate that with standard doses of both agents there is no difference in the rate of progression of CMV retinitis in AIDS patients treated with either GCV or foscarnet.45,46,87 Clinical examination tends to overestimate the time to progression compared with data based on rigorous photographic endpoints.84,86,88,89
The results of a randomized, controlled trial comparing GCV with delayed therapy using strictly graded retinal photographs demonstrated that retinitis progressed while on GCV within a median of 50.5 days, compared with the progression on delayed therapy, which occurred within a median of 15 days.46 Similarly, the results of the Studies of the Ocular Complications of AIDS (SOCA) trial demonstrated that the median time to progression of CMV retinitis while on GCV was 56 days.87
In an open study of GCV for the treatment of CMV esophagitis, among 10 evaluable patients treated with an induction regimen of 2.5 mg/kg IV q8h or 5 mg/kg IV q12h for 10 days, five persons had a good response, three had a partial response, and two had no response to therapy.53 In general, in the absence of HAART, there is a high rate of relapse of CMV esophagitis in persons who receive intermittent therapy. The decision about whether to use daily, lifelong treatment or intermittent, high-dose therapy for esophagitis due to CMV must be individualized. Maintenance therapy should be considered, particularly after a relapse (Fig. 50-5).
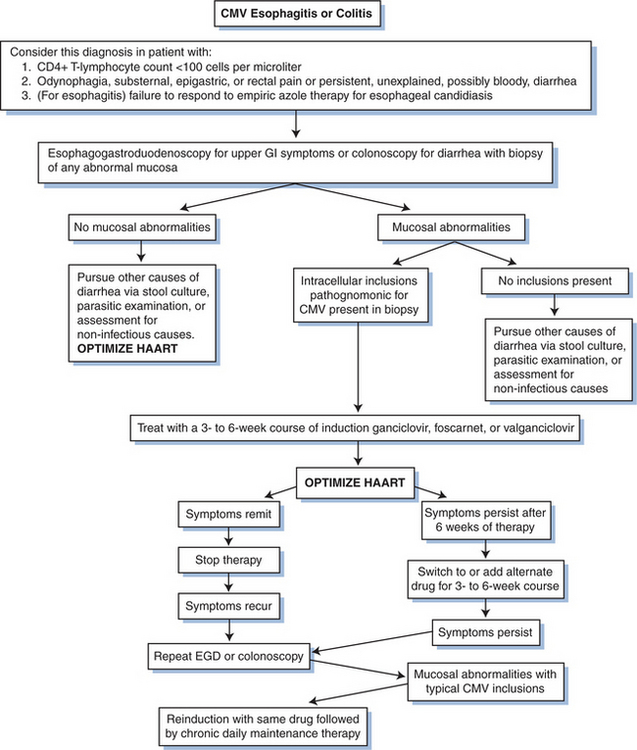
Figure 50-5 Algorithm for diagnosing and treating CMV gastrointestinal (GI) disease. EGD, esophagogastroduodenoscopy.
GCV has been evaluated in a multicenter, double-blind, placebo-controlled trial for the treatment of CMV colitis in persons with AIDS.90 Although the trial lasted only 14 days and was too short to demonstrate colonic healing and resolution of diarrhea, colonoscopy scores reflecting inflammation and positive cultures for CMV from the colon and urine significantly decreased in patients on the GCV arm. Most experienced clinicians recommend that treatment for CMV colitis should be given for 3–6 weeks. Unlike CMV retinitis, because the cells lining the gastrointestinal tract regenerate rapidly, therapy can often wait for the development of moderate to severe symptoms to justify the use of a systemic therapy with considerable toxicities. As with esophagitis, maintenance therapy is not necessarily required but should be strongly considered after a relapse. In a randomized, controlled trial of 48 patients with biopsy-proven gastrointestinal CMV disease comparing intravenous GCV (n = 22) with intravenous foscarnet (n = 26), 73% of subjects had a good or complete clinical response to a 2- to 4-week trial of either drug, with more than 83% of the subjects demonstrating an endoscopic response.91
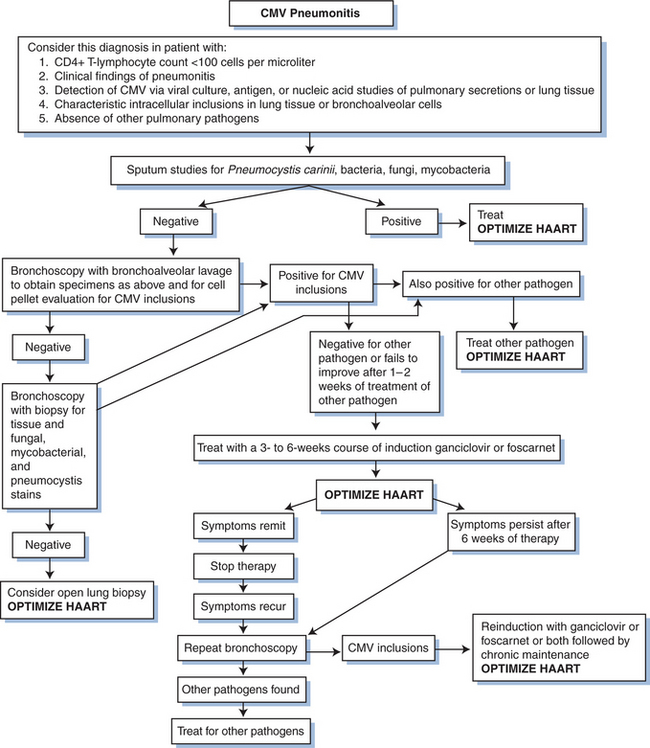
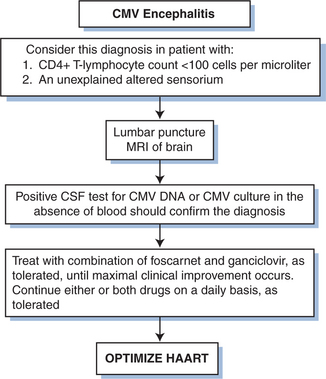
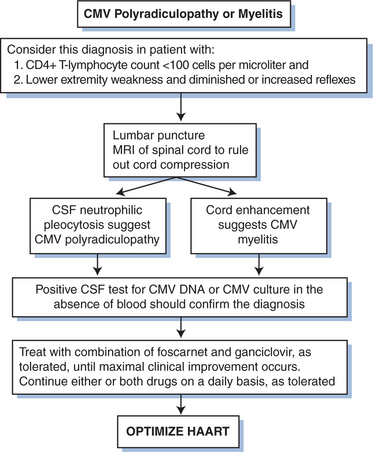
The utility of GCV therapy in the treatment of CMV in other organ systems is not proven by randomized controlled trials. Although the combination of GCV with high-dose intravenous immunoglobulin has been suggested to be more effective than GCV alone in the treatment of CMV pneumonitis in bone marrow transplant recipients using historical controls,92,93 the addition of intravenous immunoglobulin has not shown any benefit over GCV alone in persons with AIDS.94 The response of pneumonitis to intravenous GCV has been reported to be better than 60%.65 Response is probably better when patients are treated before the disease is severe. The role for maintenance therapy for CMV pneumonitis has not been established (Fig. 50-6). Many clinicians recommend such maintenance, however, as long as the CD4+ T-lymphocyte count remains below 100–150 cells/μL. For neurologic disease, initiating therapy promptly is critical for an optimal clinical response. Most clinicians treat CMV neurologic disease with GCV. Some data suggest that combination therapy with GCV and foscarnet is best for stabilizing or improving the response (Figs 50-7 and 50-8).95
Neutropenia and thrombocytopenia are the major dose-limiting toxicities of GCV therapy (Table 50-3). Because GCV and zidovudine are both myelosuppressive, it is difficult to administer these agents concurrently.96 In one study, only 18% of 29 persons with CMV disease were able to tolerate full doses of GCV with 600 mg of zidovudine daily.97 In the randomized trial comparing initial therapy with GCV versus foscarnet for the treatment of CMV retinitis, 14 of 127 (11%) patients required switching from GCV to foscarnet: nine of 14 for progression of retinitis but only one of 14 for drug toxicity.87 Limited in vitro data suggest that the antiretroviral activity of both zidovudine and didanosine may be antagonized by GCV.97
Intravitreal GCV
Intravitreal injections of GCV have been used for the treatment of CMV retinitis in persons unable or unwilling to tolerate systemic therapy with GCV or foscarnet.98–100 The concentration of GCV in intravitreal fluid immediately after injection has been as high as 65 μM.99 Concentrations higher than the ID50 for most strains of CMV can be maintained for 60h after a single 400μg injection.100 Two injections of 400μg of GCV per week are given for 2–3 weeks during an induction period, followed by weekly maintenance injections. Alternatively, a single 2 mg injection may be given weekly. This alternative therapy effectively prevents the progression of CMV retinitis without the systemic toxicity of GCV and without significant toxicity to the retina. However, adjunctive systemic therapy is recommended to protect other organs (including the contralateral eye) from viremic spread. Rarely, retinal detachment, intraocular infection, intravitreal hemorrhage, or damage to the lens occurs. One retrospective case series from the pre-HAART era reported that retinal detachment was less common among patients who received intravitreal infections compared to systemic maintenance therapy.101 This difference might be explained by high intraocular drug concentrations decreasing the area of retina involved with retinitis, so decreasing the risk of detachments.
GCV intraocular device
Local administration of GCV can be accomplished using a GCV intraocular device, licensed by the FDA for the treatment of CMV retinitis in 1996. This implant device releases GCV into the vitreous cavity at a rate of ∼1.40 μg/h for a period of 6–9 months. It is surgically implanted in the affected eye during a simple outpatient surgical procedure. Recovery time is generally of the order of several days, but persons tend to have decreased visual acuity in the implanted eye for 4 weeks.102 In a randomized, controlled clinical trial patients with peripheral CMV retinitis received either immediate implantation of the device or were closely monitored; the median time to progression of retinitis was 15 days in eyes of the delayed treatment group (n = 16) compared with 226 days in the immediate treatment group (n = 14)(p < 0.00001).102 In eight persons in whom vitreous levels of GCV were obtained, the mean vitreous drug level was 4.1 μg/mL, roughly four times the concentration in the eye obtained with intravenous GCV. The major complications of the trial included the risk of CMV retinitis development in the fellow eye (estimated at 50% at 6 months) and the development of visceral CMV disease in 31% of patients. Seven late retinal detachments occurred in this study, but because retinal detachment occurs in those with treated and untreated CMV retinitis41,103,104 it appears that an increased rate of early, surgically associated retinal detachment may be balanced by a decreased rate of late retinal detachments due to better control of the CMV retinitis.
A randomized, controlled clinical trial compared the use of intravenous GCV with that of two GCV implant devices releasing GCV at different rates.105 The median time to progression of retinitis was 191 days (71 eyes) and 221 days (75 eyes) with the two implants, compared with 71 days (76 eyes) with intravenous GCV (p < 0.001). Extraocular CMV disease occurred more commonly in persons with the implant devices than in those receiving intravenous GCV (10.3% vs 0%; P = 0.04). Vitreous hemorrhage, which was generally transient, occurred in 7.8% of eyes. Endophthalmitis was reported in three eyes. Retinal detachments were reported in 11.9% of eyes receiving implants compared with 5.1% of the eyes of persons receiving intravenous GCV. There was no indication of a significant difference in mortality between the groups on this study.105
To evaluate the efficacy of oral GCV in combination with the GCV implant to control the development of extraocular and fellow-eye CMV disease, a multicenter, three-arm randomized clinical trial comparing the use of the implant alone versus the implant with co-administration of oral GCV (1500 mg PO thrice daily) versus standard intravenous GCV was conducted.106 In 377 patients with AIDS and CMV retinitis restricted to one eye, after 6 months of treatment the development of extraocular CMV disease or CMV retinitis in the fellow-eye was 37.8%, 22.4%, and 17.9% in the three groups, respectively, a statistically significant finding comparing either the combination or the intravenous GCV groups with the implant-alone group. Progression of retinitis was also decreased by systemic therapy, consistent with the possibility that some progressions may be caused by exogenous viremic reinfections.107 It is of note that the rate of development of Kaposi sarcoma was similarly decreased, occurring in 11.3%, 2.7%, and 1.5% of the three groups, respectively. This is the first time that use of an antiviral agent in a randomized clinical trial decreased the incidence of Kaposi sarcoma. Doses of VGCV currently used include 450 mg bid and 900 mg od.

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


