Type I Interferons
The Type I IFNs consist of at least five classes in humans of which IFN
α and IFN
β have been used the most clinically.
3 Both IFN
α and IFN
β have properties that make them attractive as immunotherapies. They up-regulate major histocompatibility complex (MHC) class I molecules and induce maturation of a subset of dendritic cells (DCs).
10,
11 Type I IFNs also activate cytotoxic T cells (CTLs), natural killer cells (NK) cells, and macrophages.
12,
13,
14 Mice with targeted deletion of the Type I IFN receptor have a higher rate of carcinogen-induced cancer compared to controls and have enhanced tumor development in transplantable tumor models, supporting the hypothesis that the Type I IFNs are important in immunosurveillance.
15,
16 In addition to their immunologic effects, the Type I IFNs can have a cytostatic effect on tumors cells and may be proapoptotic.
17,
18 They also have antiangiogenic effects on the tumor vasculature.
19,
20 These observations suggest that IFNs may have direct tumor effects in addition to its immune effects. Experiments using tumors that are deficient for STAT1, a critical downstream signaling molecule of IFN, demonstrated that the presence or absence of STAT1 in the tumor had no effect on response to IFN treatment.
21 In contrast, the mice that were deficient in STAT1 that were inoculated with the STAT1-competent B16 murine melanoma cell line derived no survival benefit from the administration of IFN
α, whereas the WT mice had improved survival.
21 These data suggest that the major effect of IFN in vivo is through the host cells. This experiment does not address whether the observed effect is through immune response or inhibition of angiogenesis. Despite extensive investigation, the primary mechanism of IFN antitumor effects in humans remains to be clarified.
Type I IFN can be produced by almost all cells in the body in response to viral and bacterial nucleic acids and bacterial cell wall components such as lipopolysaccharide (LPS), but it was recognized that a small subset of peripheral mononuclear cells produce the majority Type I IFN.
22 Plasmacytoid dendritic cells (pDC) were identified as the elusive “natural type-I-IFN-producing cell.”
23,
24 On exposure to viral and bacterial products, the Toll-like receptors (TLRs) are engaged and activate multiple signaling pathways, including the NF-
κB pathway, mitogen-activated protein kinase (MAPK) pathway, and IFN regulatory factors (IRFs, discussed below), which lead to maturation of the DC and induction to the Type I IFNs (reviewed in Refs [
25,
26]). Flt3 ligand can be used to differentiate bone marrow cells into pDC and mobilize pDC into peripheral circulation in humans.
27,
28 Although the mechanism for IFN induction for infections has been well characterized, how IFN is induced in the setting of malignancy and immunosurveillance is unclear. Interestingly, the IRFs have been implicated in the DNA damage response and this may provide a mechanism by which abnormal cells initiate an innate immune response.
29,
30,
31,
32
Type I Interferon Signal Transduction
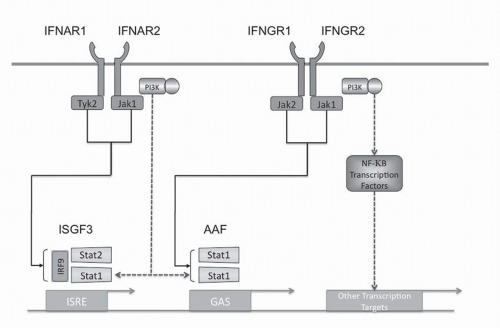
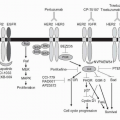
Type I IFN intracellular signaling is mediated through multiple pathways (see
Fig. 31-2). The Type I IFN receptor subunits constitutively associate with members of the Janus kinase (JAK) family. IFNAR1 associates with TYK1 and IFNAR2 associates with JAK2.
33,
34 Ligand binding results in activation of these JAK kinases as well as phosphorylation of tyrosine residues in the receptor chains. These events lead to phosphorylation and activation of members of the signal transducer and activator of transcription (STAT) family. The Type I IFN receptor has been shown to phosphorylate all seven members of the STAT family but STAT1 and STAT2 appear to be the main effectors. The phosphorylated STATs
form homodimers or heterodimers and translocate to the nucleus. Two transcriptional complexes are formed, IFN-
α-activated factor (AAF, IFN-
γ-activated factor [GAF]) and IFN-stimulated gene factor-3 (ISGF3).
35 AFF is a homodimer of phosphorylated STAT1, which binds to the IFN
γ activated sequence (GAS), and ISGF3 is a heterodimer of STAT1 and STAT2 complexed with IRF-9 (p48, ISGF3
γ) that binds to the IFN-stimulated regulatory element (IRSE).
35,
36,
37 In addition to STAT dimers, CT10 oncogene homologue (avian)-like protein (CrkL) forms heterodimers with STAT5 upon receptor activation and binds with the GAS sequence.
38 Although the JAK/STAT pathway is the classic mediator of IFN signaling, a number of other signaling pathways play a critical role in IFN signaling.
The Type I IFN receptor activates the phosphatidylinositol 3-kinase (PI3K)/AKT pathway as well as members of the MAPK family.
39,
40,
41 PI3K activation generates the phosphatidylinositols, PIP2 and PIP3.
42 These second messengers activate downstream targets such as 3-phosphoinositide-dependent protein kinase 1 (PDK1) by recruitment or conformational change.
43 PDK1 in turn phosphorylates AKT (protein kinase B) on threonine 308, resulting in activation.
43 AKT is further activated by phosphorylation on serine 473 by a protein complex containing Tor.
44,
45,
46Torc 1 and 2 are enzymatic complexes of the mammalian target of rapamycin (mTor). Torc1, the rapamycin sensitive complex, consists of mTor, Raptor (regulatory associated protein of mTor), and mLST8 (mammalian lethal with sec18 protein 8). It regulates protein translation by phosphorylating ribosomal S6 kinases, S6K1 and S6K2, and the eIF4E (eukaryotic initiation factor 4E)-binding proteins, 4E-BP1 and 4E-BP2.
44,
45,
46 Torc1 regulates translation and has been implicated in autophagy.
47 Torc2, the rapamycin insensitive complex, consists of mTor, Rictor (rapamycin-insensitive companion of mTor), mLST8 (mammalian lethal with sec18 protein 8), and mSin1 (mammalian stress-activated protein kinase-interacting protein-1). This complex phosphorylates AKT on serine 473, leading to phosphorylation and inhibition of FOXO transcription factors. Torc2 in concert with PDK1 phosphorylates and stabilizes PKC
α.44,
45,
46 The PI3K/AKT/mTor pathway regulates apoptosis, gene expression, protein translation, and cytoskeletal organization.
Activation of the Type I and II IFN receptors activates the class I PI3K, which is composed of regulatory (p85) and catalytic subunits (p110).
41,
48,
49 Experiments using MEFs from p85 knockouts have shown that PI3K plays a role in the induction of IFN-stimulated genes (ISG) including ISG15, CXCL10, and IRF by regulating mRNA transcription through Torc1.
41 In addition, PI3K plays a role in phosphorylation of STAT1 on serine 727, which enhances STATdependent transcription.
50 A relatively small number of genes that are regulated by IFN are modulated by PI3K/mTor pathway.
51 The induction of IFN in pDC is also dependent on the PI3K/mTor pathway. These findings elucidate the molecular mechanism of immune suppression by rapamycin.
52The NF-
kappaB (NF
κ B) transcription factors play a critical role in the suppression of apoptosis and regulation of the cell cycle. The NF
kgr; B family consists of p50, which is derived from the p105 precursor and p52, which is derived from the p100 precursor, RelA (p65), RelB, and cRel. The p50 and p52 subunits lack a transcription activation domain and as homodimers function as transcriptional repressors or form complexes with BCL-3 to activate transcription.
53,
54 p50 is constitutively processed from p105, but the processing of p100 to p52 is tightly regulated by phosphorylation and ubiquitinylation.
53 In contrast, p65, cRel, and RelB have a transcription activation domain and thus, when complexed with p50 or p52, are capable of activating transcription.
53 NF
κB homodimers and heterodimers are tightly regulated and are bound in the cytoplasm by inhibitors of NF
κB (I
κB). In the classical pathway of NF
κB activation, I
κB kinases (IKK) beta and gamma phosphorylate I
κB, leading to I
κB proteolytic degradation.
53 NF
κB then freely moves into the nucleus and regulates transcriptional activity. In the alternative NF
κB activation pathway, the TNF receptor-associated factors (TRAFs) activate MAP3K kinase and NF
κB-inducing kinase (NIK), which leads to the processing of p100 to p52 in an IKK
α-dependent manner.
53 The Type I IFN receptor activates NF
κB through the classical and alternative pathways.
55,
56 The classical pathway appears to be PI3K dependent, while the alternative pathway appears to be dependent on NIK.
55,
56 NF
κB binds to the promoter of a subset of IFN-induced genes that also have binding sites for IRFs and STATs.
The IRF transcription factors are intricately integrated into the regulation of IFN. Mouse knockouts have demonstrated that IRF-4 and IRF-8 are required for the generation of pDCs.
57 IRF-3 and IRF-7 along with NF
κB directly regulated the expression of Type I IFNs.
58 ISGF3 complex, which contains IRF-9, is a signaling complex downstream from the Type I receptor that induces IRF-7 expression.
59 ISGF3 and IRF-1 both bind to the ISRE DNA sequence and IRF-1 acts as a positive regulator of Type I and Type II IFN-induced genes, whereas IRF-2 represses the effects of IRF-1.
60 The induction of ISGF3, NF-
κB, and IRF-7 by the Type I IFN receptor provides a potential positive feedback mechanism for IFN in pDCs.
61,
62,
63,
64In contrast to the positive regulators of IFN signaling (e.g., IRFs), activation of negative regulators attenuates IFN signaling. Phosphatases are the counter balance to kinase activity. Src-homology 2 domain-containing protein tyrosine phosphatase-1 (SHP-1) and SHP-2 are protein tyrosine phosphatases that contain SH2 domains. SHP-1 appears to be a negative regulator of signaling with multiple reported substrates.
65 SHP-1 can dephosphorylate JAK family kinases and terminate signaling.
66 SHP-2 appears to be a positive modulator of MAPK signaling, but it may also function as a negative effector.
67,
68 Both phosphatases have been shown to have a negative regulatory role in IFN signaling as a heightened response to IFNs is observed in SHP-1- and SHP-2-deficient cells.
68,
69 In addition, treatment of cells with stibogluconate, an inhibitor of both phosphatases, resulted in prolonged IFN signaling.
70 Thus, SHP-1 and SHP-2 would be potential pharmacologic targets to modulate the effect of IFNs.
STAT signaling is regulated by suppressor of cytokine signaling (SOCS) proteins and the protein inhibitor of activated STATs (PIAS) gene families. The SOCS proteins are induced by IFN and negatively regulate cytokine signaling through binding and inactivation of JAKs and STATs, targeting the protein for degradation, or both.
71,
72,
73 The PIAS proteins bind to STAT1 and STAT3 dimers, thereby blocking their DNA-binding activity. PIAS1 selectively regulates a subset of IFN-
γ– or IFN-
β-inducible genes by interfering with the recruitment of STAT1 to the gene promoter.
74 Both these pathways likely play critical roles in dampening the response to IFN in vivo.