initiating event (activated H-RAS), albeit with a longer latency than in mice with deletions affecting the entire locus.8 Notably, the tumors in these mice were also found to harbor either deletion of ARF or mutation of p53. Therefore, while INK4A is a bona fide tumor suppressor, additional genetic dysregulation of the p53 pathway seems obligatory for melanoma genesis, at least in the mouse.
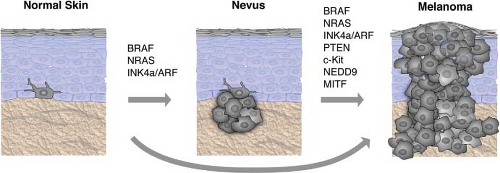 FIGURE 27.1 Validated genes mutated or deregulated in melanoma progression. Progression of normal melanocytes to metastatic melanoma is diagrammed with associated genetic events as a linear process, although in a majority of the cases, melanomas may not arise from a pre-existing nevus. The degree to which the association of each gene has been experimentally validated is shown in Table 27.1. |
TABLE 27.1 SUMMARY OF VALIDATED GENES INVOLVED IN MELANOMA GENESIS AND PROGRESSION | |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
 FIGURE 27.2 The unusual genomic structure and products of the CDKN2A locus. A: INK4A and ARF (p14ARF in human and p19ARF in mouse) initiate in different first exons and share the coding exon 2, but in an alterative reading frame, thus encoding two proteins with no amino acid similarity. The involvement of the neighboring and related CDKN2B locus in the pathology of melanoma is unclear, although it is often deleted in conjunction with CDKN2A. (From ref. 3.) B: ARF participates in the p53 pathway by binding to and sequestering the p53 antagonist, MDM2. The loss of ARF therefore results in the net degradation of p53 by MDM2-mediated ubiquitination. p16INK4A inhibits the action of the CDK4/CDK6-cyclin D1 complex in the retinoblastoma (RB) pathway. The loss of p16INK4A results in the phosphorylation and inactivation of pRB leading to its uncoupling from the transcription factor E2F, allowing for transcription of E2F target genes that are required for cell cycle progression from G1 to S phase. |
focal amplifications at the Cdk6 locus,18 which encodes an orthologue to CDK4. Furthermore, p53 heterozygous mouse melanomas retain Arf, demonstrating their epistatic relationship.16
 FIGURE 27.3 The melanoma signaling cascades mitogen-activated protein kinase (MAPK) and phosphatidylinositol 3-kinase (PI3K). The MAPK pathway is hyperactivated in melanomas, mainly due to activating mutations in either the NRAS or BRAF genes. The sequential phosphorylations of the downstream MEK and ERK proteins ultimately results in the activation of a number of transcription factors, including MITF, which induce cell proliferation and survival. The PI3K pathway is mainly hyperactivated by loss of the PTEN tumor suppressor, which results in the phosphorylation and activation of the survival gene AKT and subsequent stimulation of the mitogenic mammalian target of rapamycin (mTOR) pathway. The relative independence of the MAPK and PI3K pathways is supported by the apparent need for mutations that affect both. |
melanomas rarely, if ever, metastasize, N-RAS tumors frequently metastasize to draining lymph nodes and distal organs, in line with the apparent selection for N-RAS over H-RAS mutations in human melanomas. Knockdown of NRAS in human melanoma cell lines inhibits their viability, indicating dependency on this oncogene for tumorigenicity.25 Furthermore, shutting off transgene expression in an inducible H-RAS model caused regression of melanomas that arose following transgene induction, thereby confirming the RAS oncogene dependency in these tumors.26
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


