Major Concepts
Diseases
Prions, self-replicating, infectious proteins, are responsible for transmissible spongiform encephalopathies (TSEs) in humans and animals. The human forms include several types of Creutzfeldt-Jakob disease (CJD), Gerstmann-Straussler-Scheinker syndrome (GSS), fatal familial insomnia, Alper’s syndrome, and Huntington disease–like syndrome 1. Increased pathology may occur during Wilson disease and primary progressive aphasia. Prion diseases may be acquired by inherited mutations of the prion gene, PRNP, or by infection by malformed prions, PrPsc. The latter are transmitted by consumption of cattle or human prions or by medical procedures involving infected instruments or blood, meninges, corneas, or pituitary-derived human growth factor. Prion diseases of animals include bovine spongiform encephalopathy (BSE) (in cattle), scrapie (in sheep), chronic wasting syndrome (in deer, moose, and elk), transmissible mink encephalopathy (in mink), and feline spongiform encephalopathy (in domestic cats). Other than the BSE agent, none of the animal TSE agents appears to directly cause disease in humans.
Symptoms
Prion diseases are uniformly fatal and are accompanied by ataxia, paralysis, emotional disturbances, loss of cognition, and dementia due to the deposition of amyloid fibrils and spongiform degeneration of gray areas of the central nervous system. The cerebrum, cerebellum, basal ganglia, and thalamus may be affected.
Infection
The human prion protein exists in two forms. The normal form, PrPc, is found on the surface of neurons, glial cells, platelets, and various leukocytes; is protease-sensitive; and contains large amounts of α-helical secondary protein structure. The abnormal form, PrPsc, may be found in lysosomes within infected cells, is protease-resistant, and is aberrantly folded, containing large amounts of β-pleated sheets in its secondary structure. PrPsc is formed by interacting with PrPc and refolding the normal protein into PrPsc, with more β-pleated sheet content. Prions are thus able to replicate themselves without containing or using nucleic acids. The exact function of the normal PrPc is not known. Mice lacking this protein appear to be grossly normal but have alterations in circadian rhythms, sleep patterns, synaptic conduction, seizure thresholds, and hippocampus morphology. PrPc may be involved in copper transport, immune cell functioning, and inhibition of the processing of the Aβ amyloid protein found in Alzheimer’s disease. In addition to mammalian prions, six prions have been found in fungi.
Immune Response
The immune system is activated during prion diseases because infection stimulates the release of T cell–tropic chemokines followed by the entry of CD4+ T helper cells and CD8+ T killer cells into the brain tissue. These cells do not appear to mount a protective response, however, and the disease proceeds until death. B lymphocytes appear to play a contributory role in disease progression. Leukocytes in the intestinal wall, as well as the cytokines TNF-α and TNF-β, may be required for infection by the oral route.
Protection
No treatment has thus far been able to stop the fatal progression of prion diseases in humans, although several drugs may slow the course of disease in animal models. Research is continuing to uncover new drugs that cure neuronal cells in culture, but delivery of these agents in vivo is complicated by the blood-brain barrier. Several efforts have been able to reduce or prevent transmission. Since humans may be infected by eating beef or other meat from BSE-infected cattle, a number of ruminant feed bans have been put into effect that have greatly reduced the incidence of BSE in cattle and the resultant variant Creutzfeldt-Jakob disease in humans. Kuru, another form of CJD, has been almost eliminated by halting funeral rites that involved consumption of human brains. Incidence of iatrogenic CJD is reduced by disposing of infected medical instruments and refusing blood from at-risk donors, such as those who have received a meningeal or corneal transplant, are receiving pituitary-derived human growth hormone, or have lived for more than six months in regions with a high incidence of BSE.
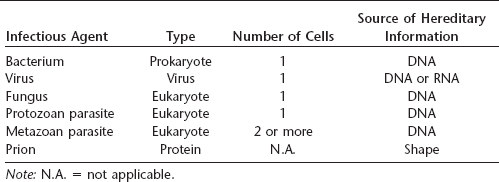
Almost all infectious agents fall into several well-defined categories, including bacteria, viruses, fungi, and protozoan (unicellular) or metazoan (multicellular) parasites. All of these agents are considered by at least some authorities as alive (although others do not classify viruses as such), and all use a nucleic acid, either DNA or RNA, as their hereditary information. Prions are different from any other known infectious agent: they are not living and are composed solely of a single protein without any nucleic acid. Prion hereditary information used during reproduction appears to be contained in the shape of the protein. These agents may pass from one individual to another via an infectious route or may be transmitted to offspring through an inherited mutation in the gene that encodes a normal cellular form of the protein.
Table 28.1 Infectious agents of humans
Prion proteins are found primarily on the surface of neurons but are also present on glial cells, platelets, and some leukocytes, including hematopoietic stem cells. The exact function of the normal protein is not clear, but it may function in copper ion transport, in blocking the formation of amyloid plaques associated with Alzheimer’s disease, during T lymphocyte activation, during blood cell production, or in synaptic transmission. These unusual infectious agents cause several universally fatal diseases of the central nervous system, characterized by loss of coordination, inability to move, emotional disturbances, dementia, and finally death. “Mad cow disease” is one of these transmissible spongiform encephalopathies that infects cattle but may be transmitted to humans by consuming infected beef. Because prions are not inactivated by extremely high or low temperatures, normal cooking or freezing procedures do not block transmission. Iatrogenic transmission may also occur because normal decontamination procedures, including autoclaving, do not inactivate these hardy infectious agents.
Scrapie, a transmissible spongiform encephalopathy (TSE) of sheep, was reported in 1730 in Europe, and in 1936 it was found to be transmissible between sheep. The first of the human TSEs was identified by Hans Gerhard Creutzfeldt and Alfons Maria Jakob in the 1920s and was named Creutzfeldt-Jakob disease (CJD). Daniel Carleton Gajdusek described kuru among the cannibalistic Fore tribesmen of Papua New Guinea in 1957. Gajdusek received a Nobel Prize in 1976 for his work, which proved that the prions that caused kuru and CJD were transmissible. In 1966, Tikvah Alper found that the scrapie agent was extremely resistant to inactivation by ultraviolet light and questioned whether it could replicate without nucleic acids. Prions, the self-replicating, infectious proteins responsible for TSEs in humans and animals, were purified in 1982 by Stanley Prusiner, who received the Nobel Prize in Medicine in 1997 for this work.
Variant Creutzfeldt-Jakob disease (vCJD), the human form of “mad cow disease,” was initially noted when ten persons with an atypical form of CJD were diagnosed between February 1994 and October 1995 in widely dispersed areas of the United Kingdom. This disease is linked to human consumption of beef originating in cattle from the United Kingdom that had bovine spongiform encephalopathy (BSE). During the 1970s, changes were made in the methods for rendering sheep offal used in cattle feed by abandoning the use of organic solvents. Some of the sheep offal was derived from animals with scrapie, a spongiform encephalopathy of sheep that does not directly infect humans. Meat and bone meal from BSE-infected cattle was also included in the feed, amplifying the epidemic. By 1988, more than 168,000 cases of BSE had been reported in the United Kingdom, and the number of vCJD cases in humans was also rising. In an attempt to halt the outbreak, a ban on feeding ruminant-derived protein to other ruminants went into effect in July 1988. Between November 1989 and January 1990, this ban was expanded to preclude the use of cattle brain, spinal cord, tonsil, thymus, spleen, and lower intestine from animals over the age of 6 months in human foods; beginning in September of that year, the ban was extended to include feed for all animals and birds. “Beef on the bone” was later banned as well. Milk and milk products, however, are considered safe for human consumption. The outbreak in cattle is decreasing due in part to these measures: whereas 1,000 new cases were reported per week in 1993, the number had been reduced to 300 per week in 1996.
Prions are responsible for causing TSEs, fatal diseases in which areas of spongiform (spongelike, full of small holes) degeneration develop in gray areas of the brain and spinal cord. Vacuolation of the neuropil and astrocytosis (increased numbers of the astrocyte type of glial cell) may be present. Disorders of the pancreas may also occur. TSEs may be transmitted to laboratory animals by injecting material derived from affected brains. In addition to humans, TSEs have been reported to affect cattle, sheep, goats, mice, mink, deer, and cats.
Creutzfeldt-Jakob Disease (CJD)
Persons suffering from this group of uniformly fatal diseases demonstrate loss of motor control, including rapid jerky movements, progressive dementia, paralysis, wasting, and death typically within 3 to 12 months. The cerebellum, basal ganglia, and lower motor neurons are often affected.
Sporadic CJD
Approximately 85% of the cases of CJD are sporadic, in which the means of disease acquisition is unknown. The incidence of sporadic CJD is one case per million persons per year. It primarily affects people between the ages of 50 and 75 years. This form of CJD has cerebral involvement associated with rapidly progressive dementia.
Familial CJD
Familial CJD accounts for 10% to 15% of the cases of CJD. Disease is linked to a hereditary mutation of the gene encoding the prion protein. It is inherited as an autosomal dominant trait, meaning that only one mutated copy of the gene is required to cause disease. Familial clusters have been reported in Slovakia, Israel, and Chile.
Iatrogenic CJD
Fewer than 5% of the cases of CJD are believed to be of iatrogenic origin (associated with medical procedures). Transmission of this form of disease occurs through contaminated surgical instruments, such as scalpels and cortical electrodes, via transplantation of brain meninges or corneas, or by the use of human growth hormone derived from human pituitaries. Prion transmission by blood transfusion or organ donation may also occur. Accordingly, the American Red Cross restricts blood donation by individuals who have been recipients of meningeal transplants, pituitary-derived human growth hormone, or non-U.S. licensed bovine insulin as well as those who have spent extended periods of time in the United Kingdom, due to the risk of consuming contained beef by persons in the latter group.
Kuru
Kuru, also known as “laughing disease,” was found only among women and children of the Fore Highlander tribe of Papua New Guinea. It was acquired by handling, preparing, or ingesting contaminated human brains. Fore Highlanders formerly honored their dead relatives by eating their brains. After this practice was halted, kuru almost disappeared and is now found in fewer than ten persons per year.
Variant CJD
Variant CJD (vCJD; formerly new variant CJD) is the human form of bovine spongiform encephalopathy and appears to be acquired by ingestion of contaminated beef. This form of CJD affects younger people than does sporadic CJD; the average age of onset is 27 years. The duration of illness is also longer for vCJD than for other forms of CJD (14 months versus 4½ months). Most cases of vCJD have been acquired in the United Kingdom or Ireland; a lower number were acquired in France and other parts of Europe. The incidence of vCJD has been decreasing as infected or potentially infected cattle are prevented from entering the human food chain.
Several differences exist between the clinical presentation of sporadic CJD and vCJD: patients with the former have more prominent dementia and early onset of neurological signs, whereas those with the latter have more delayed neurological signs, painful dysesthesias, prominent psychiatric symptoms such as depression or psychosis, and memory impairment. Later during the course of the disease, early ataxia (unsteadiness and loss of coordination) and involuntary movements appear; prior to death, affected individuals become completely immobile and mute. The disease is accompanied by extensive deposition of prion proteins (PrP) and amyloid plaques in the cerebellum (the region of the brain controlling balance, posture, and coordination of fine movements). Numerous “florid plaques” and the “pulvinar sign” are often present in vCJD as well. A florid plaque consists of a central area that stains with the red dye eosin and is surrounded by a region of spongiform degeneration, while the pulvinar sign is the presence of a high intensity signal from the pulvinar region of the thalamus that is detected by magnetic resonance imaging. Findings in electroencephalograms also differ between sporadic and variant CJD: the former often has periodic sharp waves that the latter typically lacks. In vCJD but not sporadic CJD, prions are typically detected in lymphoid organs. In vCJD, the spongiform changes are most evident in the brain’s basal ganglia and thalamus. The basal ganglia regulate voluntary motor activity, while the thalamus serves as the major integrating and relay station for sensory impulses. Prions accumulate in high density in the cerebellum.
Variant CJD is caused by human infection with the BSE (“mad cow”) agent. The causative agent of vCJD contains a molecular marker that is absent in other human CJDs but is present in BSE. Its transmission characteristics are almost identical to those of BSE. Evidence from animal models also supports the linkage between vCJD and BSE. Transgenic mice containing human genes are susceptible to BSE. Rhesus macaques injected with BSE material also develop a disease similar to vCJD in humans.
Table 28.2 Differences between sporadic and variant CJD
Source: CDC
| Characteristic | Sporadic Cjd | Variant Cjd |
| Median age at death | 68 years | 28 years |
| Median duration of illness | 4 to 5 months | 13 to 14 months |
| Clinical signs and symptoms | Dementia; early neurological signs | Prominent psychiatric and behavioral symptoms; painful dyesthesiasis; delayed neurological signs |
| Periodic sharp waves on electroencephalogram | Often present | Often absent |
| “Pulvinar sign” on MRI | Not reported | Present in more than 75% of cases |
| Presence of “florid plaques” on neuropathology | Rare or absent | Present in large numbers |
| Immunohistochemical analysis of brain tissue | Variable accumulation of protease-resistant prions | Marked accumulation of protease-resistant prions |
| Presence of prion in lymphoid tissue | Not readily detected | Readily detected |
Gerstmann-Straussler-Scheinker Syndrome
Gerstmann-Straussler-Scheinker syndrome (GSS), like familial CJD, is caused by an inherited autosomal dominant mutation to the prion gene but may rarely occur spontaneously as well. It generally affects persons in their forties or fifties. GSS leads to cerebellar ataxia; dementia is seen less commonly than in CJD. Multicentric amyloid plaques are common. This condition typically persists for several years before terminating in death.
Fatal Familial Insomnia
Fatal familial insomnia (FFI) is a form of human TSE that involves the autonomic nervous system. It is characterized by progressive disturbances of sleep, blood pressure, body temperature, and appetite due to severe atrophy of the thalamus of the brain. Little involvement of the neuropil occurs during FFI. This disease is usually diagnosed among persons between 40 and 60 years of age, who generally live 7 to 18 months postdiagnosis.
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


