Known variously as
disseminated intravascular coagulation (DIC),
defibrination, or the
consumption coagulopathies, consumptive thrombohemorrhagic disorders are a heterogeneous group of disorders that can be manifested by the entire range of hemorrhagic and thrombotic pathology. Essentially, DIC comprises a clinicopathological state in which systemic activation of coagulation occurs, leading to thrombotic obstruction of small-and mid-size vessels
1,2,3,4,5 impairing blood supply and contributing to multiple-organ dysfunction (MOD). Due to ongoing activation of the coagulation system and other processes, such as impaired synthesis of platelets and coagulation proteins, depletion of coagulation factors, protease inhibitors, and platelets may occur.
6,7 This situation leads to a severe impairment of hemostasis and results in serious bleeding, in particular in patients who are at risk for major blood loss, such as perioperative patients or trauma patients. In fact, bleeding may dominate the clinical picture of a patient with DIC.
In view of these multiple, often contrasting mechanisms that occur in patients with DIC, a consensual definition of DIC had been a matter of debate. In 2001, the subcommittee on DIC of the International Society on Thrombosis and Hemostasis proposed a definition that reflects the central role of the microvascular milieu, that is, endothelial cells, blood cells, and the plasma protease system, in the pathogenesis of DIC. This definition of DIC reads as follows: “DIC is an acquired syndrome characterized by the intravascular activation of coagulation without a specific localization and arising from different causes. It can originate from and cause damage to the microvasculature, which if sufficiently severe, can produce organ dysfunction.”
8The diagnosis of DIC may be hampered by the nonspecific nature of many indicators of coagulation activation, although newly developed scoring algorithms, based on readily available routine laboratory parameters, show promising diagnostic accuracy.
8,9 Owing to the complexity of the clinical presentation, the variable and unpredictable course, and the multitude of therapies given to patients with DIC, properly conducted clinical trials are extremely difficult to perform and even to devise. Management relies on limited evidence from clinical trials in combination with small studies employing surrogate outcome parameters and experience from case series, as well as an understanding of the pathophysiologic mechanisms involved, together precariously applied to the individual patient.
10
HISTORY
One of the first reports of DIC comes from Dupuy in 1834, who described the effect of the intravenous injection of brain material in animals.
11 The animals almost immediately died, and at autopsy there were widespread clots in the circulation, presumably due to what we would now call tissue factor (TF)-dependent systemic activation of coagulation. Thirty years later, Trousseau described the thrombotic tendency and the inclination of blood to clot in patients with advanced malignant disease.
12,13 Nauyn observed gross intravascular coagulation of animal blood after infusion of hemolyzed erythrocytes,
14 and Foa and Pellacani
15 observed the same phenomenon after injection of a variety of fresh organ extracts. Mellanby recognized that the coagulation defect of DIC produced by snake venoms resulted in hypofibrinogenemia.
16,17 A more precise description of DIC and its underlying pathogenesis had to wait until 1955 when more insight into the mechanism of blood coagulation was attained and better laboratory tests had become available. Ratnoff et al.
18 describe the hemostatic abnormalities that occur in women with fetal death and amniotic fluid embolism. McKay
19 was the first to realize that DIC was an “intermediary mechanism” in many diseases. Hardaway
2 emphasized that shock and acidosis occurring in a variety of diseases could initiate clotting with consumption of fibrinogen, platelets, and other coagulation factors. Verstraete et al.
20 reported several other cases of successful heparin use in DIC and suggested that this drug might be of more general value.
CLINICAL FEATURES OF CONSUMPTIVE THROMBOHEMORRHAGIC DISORDERS
The spectrum of clinical and laboratory presentation is affected by several parameters, as listed in
Table 98.1. Herein lies much of the confusion, especially because sometimes cases of so-called DIC are not disseminated, or intravascular, or even related to coagulation. These variables determine whether specific (supportive) therapy for the “consumption” is required, over and above that indicated for the underlying disease or its other complications.
Table 98.2 summarizes the clinical manifestations of severe consumptive hemorrhagic disorders.
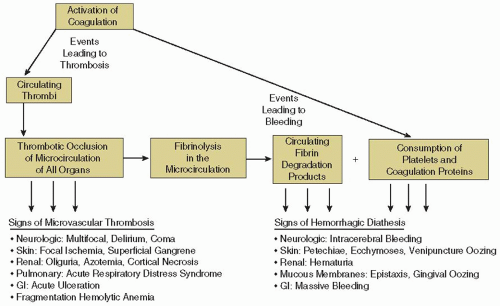
When presented with the patient with a clinical suspicion of DIC, the physician must judge whether the clinical presentation is severe enough to warrant specific therapy, and whether the coagulation disorder is likely to be a self-limiting problem that will wane on therapy for the underlying condition or will gather momentum and assume importance beyond that of the initiating stimulus. If the problem comes to the attention only as the result of a screening laboratory survey, management will depend on whether an abnormal test result is the only reflection of consumption or whether serious clinical manifestations also exist or are likely to develop. In particular, signs of ischemic organ dysfunction, macrovascular thrombosis, excessive bleeding, and fragmentation hemolytic anemia should be sought (
FIGURE 98.1).
Given that factors of tempo, location, and pathophysiology can all operate as independent variables superimposed on a variety of underlying disorders, each patient’s clinical and laboratory presentation must be individually considered before rational management can be started.
Many clinical entities have been associated with DIC,
1,21 and the major conditions are listed in
Table 98.3. The frequency of DIC in these diseases varies considerably, ranging from rare or doubtful according to current criteria for DIC to highly frequent (severe gram-negative sepsis). Infectious diseases and malignant disorders together account for approximately two-thirds of DIC cases in the major series.
22,23,24 Trauma was a major cause of DIC in some series, probably reflecting the specialized nature of the clinical case mix in those centers.
25
ACUTE, SEVERE CONSUMPTION COAGULOPATHY
Acute, severe DIC could be defined as a “pathological syndrome resulting from activation (and consumption) of certain coagulant proteins, formation of thrombin, and production of fibrin thrombi.”
7 Diffuse multiorgan bleeding, hemorrhagic necrosis, microthrombi in small blood vessels, and thrombi in medium and large blood vessels are common findings at autopsy, although patients who had unequivocal clinical and laboratory signs of DIC may not have consistent postmortem findings.
26,27 Conversely, some patients in whom clinical and laboratory signs were not diagnostic of DIC had typical autopsy findings.
28,29 This occasional lack of correlation among clinical, laboratory, and pathologic findings is partly due to extensive postmortem changes in the blood, for example, excessive fibrinolysis, but remains unexplained in most instances.
27 Organs most frequently involved by diffuse microthrombi are the lungs and kidneys, followed by the brain, heart, liver, spleen, adrenal glands, pancreas, and gastrointestinal organs. Most thrombi consist of fibrin monomers or polymers in combination with platelets, and activated mononuclear or other inflammatory cells and other signs of inflammatory activation.
30 In cases of long-lasting DIC, organization and endothelialization of the microthrombi are often observed. Acute tubular necrosis is more frequent than renal cortical necrosis.
26 Clinically, thrombotic occlusive events occur first, as the result of microthrombi of fibrin or platelets
that obstruct the microcirculation of organs (
FIGURE 98.1).
5 These thrombi result from clots that form either in the circulation or
in situ in arterioles, capillaries, or venules. Circulatory obstruction produces organ hypoperfusion and even ischemia, infarction, and necrosis. The process is disseminated throughout the microcirculation; therefore, all organs are potentially vulnerable. Venous thromboembolism or arterial embolism from nonbacterial endocarditis can occur but are much more common in patients with subacute or chronic DIC, particularly in those patients with malignant disease.
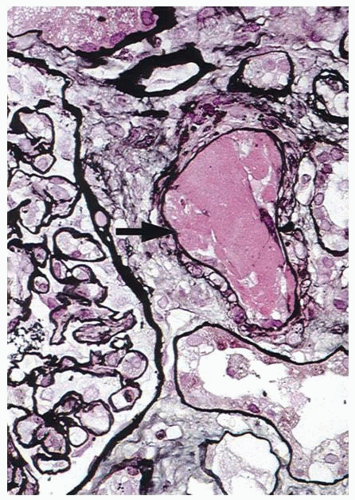
23Renal dysfunction in patients with DIC is usually multifactorial. The conditions usually associated with DIC are frequently complicated by hypovolemia or hypotension that causes prerenal azotemia, even renal failure due to acute tubular necrosis. Thrombosis of renal afferent arterioles or glomerular capillaries by fibrin thrombi may cause ischemic renal cortical necrosis (
FIGURE 98.2).
Cerebral dysfunction is most often manifested as global changes, such as an altered state of consciousness, convulsions, or coma, rather than by isolated focal lesions. Pathologic lesions affecting cerebral function may also include major vessel occlusion and subarachnoid hemorrhage, as well as multiple cortical and brainstem hemorrhages following microvascular occlusions.
31 Microthrombi, macrothrombi, emboli, and hemorrhage in the cerebral vasculature all have been held responsible for the nonspecific neurologic symptoms and signs displayed by patients with DIC
32Respiratory dysfunction in DIC can manifest as transient hypoxemia in mild cases to hemoptysis, dyspnea, and chest pain caused by pulmonary hemorrhage and adult respiratory distress syndrome (ARDS) in severe cases.
33,34,35 Physical examination reveals rales, wheezing, and occasionally pleural friction rub, and imaging shows diffuse infiltration resulting from intra-alveolar hemorrhage. ARDS is characterized by tachypnea, auscultatory silence, hypoxemia, low lung compliance, normal wedge pressure, and “white lungs” on chest images.
36 It stems from severe damage to the pulmonary vascular endothelium, which permits egress of blood components into the pulmonary interstitium and alveoli. This situation leads to intra-alveolar hyaline membrane formation and severe respiratory insufficiency.
37 ARDS can be caused by septic shock, severe trauma, fat embolism, amniotic fluid embolism, and heat stroke, but only a fraction of patients with ARDS exhibit signs of DIC. When DIC and ARDS are simultaneously triggered, each aggravates the other.
Hepatocellular dysfunction sufficient to cause jaundice has been reported in 20% to 50% of patients with DIC,
4,25 notably in patients with infection and prolonged hypotension. Cellular necrosis around the hepatic central veins may follow shock and hypoxemia or intravascular coagulation,
26,38 leading to decreased synthesis of procoagulant and anticoagulant factors.
Adrenal cortical failure, producing the Waterhouse-Friderichsen syndrome,
39 is a consequence of hemorrhagic necrosis.
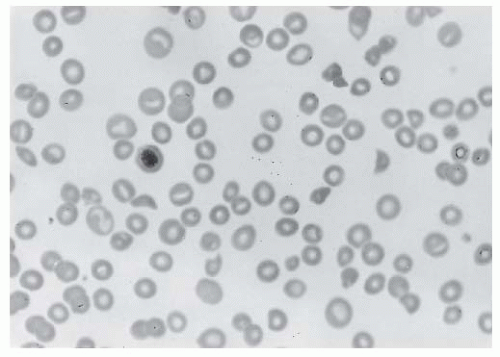
Fragmentation-type hemolysis (microangiopathy) secondary to fibrin deposition in the microvasculature may contribute to anemia
40 (
FIGURE 98.3). However, overt hemolytic anemia associated with DIC occurs in only 5% to 10% of patients, and similar morphologic abnormalities occur in many other disorders not associated with fibrin formation (e.g., thrombotic thrombocytopenic purpura [TTP]).
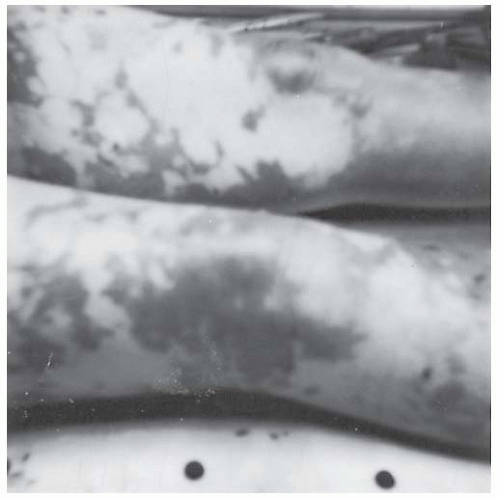
Dermal ischemia and hemorrhagic skin necrosis occurs particularly in patients with acute DIC (see
FIGURE 98.4), particularly those associated with severe infection, hypotension, and peripheral vasoconstriction. This complication of acute DIC is in contrast to idiopathic purpura fulminans, which typically occurs in young children who may be convalescing from an infection involving the skin and who are
not acutely infected at the time of the appearance of purpura fulminans.
41,42Although the initial pathologic events are manifested by fibrin deposition in the microcirculation, the patient’s initial clinical events may relate to hemorrhagic manifestations such as mucosal oozing, spontaneous ecchymoses, petechiae, and massive gastrointestinal (GI) blood loss. If the patient has had recent invasive or operative procedures, the incisions will also be vulnerable to excessive bleeding. The latter results from ongoing consumption and relatively insufficient synthesis of platelets and hemostatic factors.
Disorders Associated with Acute DIC
Infections
Virtually every type of infectious agent has been implicated in the consumptive thrombohemorrhagic disorders, usually producing mild or severe systemic intravascular coagulation, with consumption of platelets and soluble clotting factors (see also
Chapter 123:
Thrombohemorhαgic Complications of Sepsis). The most flagrant examples are the Waterhouse-Friderichsen syndrome and purpura fulminans, both of which bear pathologic and histologic similarity to the experimental animal model of consumption, the Shwartzman reaction.
43 The Waterhouse-Friderichsen syndrome is most commonly seen during fulminant meningococcal sepsis, although other organisms such as
Pneumococcus may
also cause this complication.
42,44 Widespread intravascular fibrin and platelet thrombi obstruct arterioles, capillaries, and venules of vital organs, especially causing bilateral hemorrhagic necrosis of the adrenals. This type of sepsis is associated with severe shock, which contributes significantly to high mortality. Although these clinical entities appear to be distinctive syndromes, they can be considered extreme manifestations of acute DIC. Variations in the acuteness of onset and the degree of consumption, perhaps related to the patient’s age and health and the type of infectious agent, account for the wide range of clinical manifestations, including in some patients with laboratory abnormalities of consumption but without clinical signs.
Bacterial infections are among the most common causes of DIC.
1,45 Certain patients are particularly vulnerable to infectioninduced DIC, such as immune-compromised hosts; asplenic patients whose ability to clear bacteria, particularly pneumococci, is impaired; and newborns whose coagulation inhibitory systems are immature. In addition, infections can aggravate bleeding and thrombosis by directly inducing thrombocytopenia, hepatic dysfunction, and shock.
46 Clinically overt DIC may occur in 30% to 50% of patients with gram-negative sepsis
47,48 and is common in patients with gram-positive sepsis.
49,50 Extreme examples of sepsis-related DIC are
Streptococcus A toxic shock syndrome, characterized by deep-tissue infection and vascular collapse,
51 and meningococcemia characterized by extensive hemorrhagic necrosis and shock. The extent of hemostatic derangement in patients with meningococcemia correlates with prognosis.
52,53 Patients affected by
Pseudomonas aeruginosa, Escherichia coli, and
Proteus vulgaris bacteremias may have only laboratory signs of activated coagulation or may present with severe DIC, especially accompanied by shock.
54,55The mechanism by which DIC is elicited in
Staphylococcus aureus bacteremia may be related to an α-toxin that activates platelets and induces IL-1 secretion by macrophages.
56 Streptococcus pneumoniae infection has been associated with the Waterhouse-Friderichsen syndrome particularly in asplenic patients,
57 ascribed to a capsular antigen and to antigen-antibody complex formation.
58 Clostridial bacteremia is a highly lethal disease characterized by septic shock, DIC, renal failure, and hemolytic anemia.
59Activation of the coagulation system has also been documented for nonbacterial pathogens, that is, viruses causing hemorrhagic fevers,
59,60 protozoa (Malaria),
61,62 and fungi.
63 Common viral infections, such as influenza, varicella, rubella, and rubeola, have rarely been associated with DIC.
64 However, purpura fulminans associated with DIC has been reported in patients with infections and either hereditary thrombophilias
65,66 or acquired antibodies to protein S.
67 Other viral infections can cause “hemorrhagic fevers” characterized by fever, hypotension, bleeding, and renal failure (see
Chapter 123). Laboratory evidence of DIC can accompany Korean, rift valley, and dengue-related hemorrhagic fevers,
68,69,70 perhaps secondary to TF release from cells in which viruses replicate
71 and increased levels of proinflammatory cytokines.
58
Purpura Fulminans
Purpura fulminans is a severe, often lethal form of DIC in which extensive areas of the skin over the extremities and buttocks undergo hemorrhagic necrosis.
41 The disease affects infants and children predominantly
72,73 and may occur in patients with acute bacterial infections and in patients with preexisting inherited or acquired disorders of the protein C anticoagulant pathway. Sepsis-associated purpura fulminans is much more common in children than adults,
39 Meningococcus being the most common (but not the sole) organism involved.
4 Onset may appear 2 to 4 weeks after a mild infection such as scarlet fever, varicella, or rubella or can accompany an acute viral or bacterial infection in patients with thrombophilia affecting the protein C pathway
52,72 The initial presentation in sepsis-associated purpura fulminans is often that of overwhelming acute infection with hypotension and peripheral hypoperfusion; in contrast, in idiopathic purpura fulminans, the blood pressure is well preserved. The skin necrosis in sepsis-associated purpura fulminans often begins in the distal extremities (
FIGURE 98.4), with subsequent proximal progression, or involves the entire body in a patchy distribution.
74 Infants who are homozygous for protein C or S deficiency may be born with skin necrosis and intravascular coagulation (hereditary purpura fulminans).
75,76 Patients who are heterozygotes for protein C or S deficiency may develop skin necrosis when placed on warfarin therapy
77 Protein C or S levels may be low because of liver dysfunction, consumption, or, rarely, secondary to an acquired inhibitor.
78
Trauma or Massive Tissue Necrosis
The acute coagulopathy accompanying severe trauma may complicate severe head injury, massive soft tissue trauma, multiple fractures and fat embolism, gunshot wounds, and extensive burns.
79 In a study of 16 patients with head injury who had DIC and came to autopsy within 4 days of injury, Kaufman et al.
80 noted necrosis and bleeding, most notably in the brain and lungs, and microthrombi in the central nervous system, liver, lungs, kidneys, and pancreas. Thrombi were less prevalent with
longer delays after injury, attesting to a probable fibrinolytic response in the microcirculation to dissolve these thrombi. Head injury in children is also associated with abnormal clotting tests in 71% of cases, and mortality is fourfold greater in patients with DIC than in those without DIC.
81 The high mortality rate is associated with increased bleeding and laboratory evidence of DIC and is the foundation for prognostic scoring systems.
81,82,83Extensive exposure of TF to blood and hemorrhagic shock probably are the immediate triggers of acute coagulopathy, but cytokines likely play a pivotal role. In fact, the changes in cytokine levels are virtually identical in trauma patients and septic patients.
84 The levels of TNF-α, IL-1/3, plasminogen activator inhibitor-1 (PAI-1), circulating TF, plasma elastase released from neutrophils, and soluble thrombomodulin all can be elevated in patients with signs of DIC, predicting multiple-organ failure (MOF) (ARDS included) and death.
85,86 Careful monitoring of laboratory signs of DIC, reduced fibrinolytic activity, and perhaps low antithrombin (AT) levels also are useful for predicting the outcome of such patients.
87The coagulopathy can be aggravated in patients with severe trauma who require massive blood replacement because stored blood components are diluted and do not contain sufficient amounts of viable platelets and factors V and VIII. Moreover, activation of fibrinolysis may aggravate bleeding and hypotension.
88,89,90,91The time interval between trauma and medical intervention correlates with the development and magnitude of DIC. Experience during wars proved that fast evacuation and prompt medical care reduce the risk of DIC.
92,93,94TF exposed to blood at sites of burned tissue, the systemic inflammatory response syndrome induced by the burn, and superimposed infections can trigger DIC.
95 Bleeding, laboratory tests indicative of DIC, and vascular microthrombi in biopsies of undamaged skin have been described in patients with extensive burns.
96 Significant local consumption of fibrinogen and platelets occur in burned areas.
97 Laboratory signs of DIC are associated with organ failure, and the extent of protein C and AT deficiencies correlates with poor outcome.
96 A clinicopathologic study of 139 patients who died during treatment for a severe burn disclosed that 18% had cerebral infarctions caused by septic arterial occlusions or DIC and 4% had intracranial hemorrhage.
98
Heat Stroke
Heat stroke is frequently complicated by consumption coagulopathy.
99,100 James Wellstead published in 1841 his book “Travels to the City of the Caliphs” (currently known as Baghdad) and vividly described that on an extremely hot day in the Persian Gulf the decks of the ship Liverpool resembled a slaughterhouse, so numerous were the bleeding patients.
101,102 Heat stroke is a syndrome characterized by a rise in body temperature to over 42°C that follows collapse of the thermoregulatory mechanism. Predisposing factors include high environmental temperature, strenuous physical activity, infection, dehydration, and lack of acclimatization.
103,104 Extensive hemorrhage, unclottable blood, and venous engorgement were found as early as 1841 in postmortem examination of patients who died of heat stroke.
102 Investigations have confirmed that a severe hemorrhagic diathesis and MOF often accompany heat stroke.
95,105,106,107 Diffuse fibrin deposition and hemorrhagic infarctions are found in fatal human cases. DIC associated with profound fibrin(ogen)olysis is evident in patients with heat stroke. The possible triggers of DIC in patients with heat stroke include endothelial cell damage and TF released from heat-damaged tissues.
105In a series of 18 critically ill patients from Paris with heat stroke during the 2003 heat wave in Western Europe,
107 patients showed very high levels of IL-6 and IL-8, a striking activation of white blood cells (β2-integrin upregulation and increased reactive oxygen species) and evidence of systemic activation of coagulation. There was a correlation between the extent of inflammation and coagulation activation and clinical severity of the heat stroke. Patients have been categorized as nonbleeders, bleeders without DIC but with slight consumption of hemostatic factors, and bleeders with typical signs of DIC.
99 Prompt cooling and support of vital functions have substantially reduced the high mortality that was commonly observed in early studies.
108
Snakebite
The bite of certain snakes, especially vipers and rattlesnakes, causes a coagulopathy that is highly reminiscent of DIC, although it is a matter of debate whether this condition can be truly classified as DIC.
17 Prominent among these species are the
Viperα, Echis (E. cαrinαtus or
E. colorαtus),
Aspis, Crotαlus, Bothrops, and
Agkistrodon. Venoms of these snakes contain enzymes or peptides that exert a variety of activities
109,110,111: (a) thrombin-like activity, cleaving fibrinopeptide A from the Aα chain of fibrinogen, (b) activation of prothrombin even in the absence of calcium ions, (c) activation of factors ×X and V, (d) fibrinogenolysis, (e) thrombocytopenia by platelet aggregation, (f) inhibition of platelet aggregation, (g) activation of protein C, and (h) endothelial damage cells. Victims of snake bites rarely experience excessive bleeding or thromboembolism, in spite of the serious derangements in hemostasis and findings that are sometimes consistent with DIC.
112,113,114Several species of snakes belonging to the Viperidae family produce venoms that have a wide range of activities affecting hemostasis. The symptoms are characterized by local tenderness and swelling, mild bleeding from the wound or from venipunctures, occasional hypotension without shock, and transient mild oliguria. The major symptoms and signs related to envenomation are vomiting, diarrhea, apprehension, hypotension, local swelling, ischemia, and necrosis. Treatment for victims of snake bites consists of immediate immobilization, administration of antivenom and fluids, and other general measures to preserve vital functions. Local incisions, cooling, and application of tourniquet should be avoided.
109 Some patients begin to recover spontaneously after 1 to 2 days and are completely normal within 1 week, whereas others demonstrate incoagulable blood for more than 3 weeks.
114,115 Active secondary fibrinolysis accounts for the rapid recovery of renal microcirculatory obstruction. Chugh et al.
116 documents severe illness after viper bites, with 45 of 157 patients having acute renal failure, and death occurring in eight of 10 with bilateral renal necrosis and in four of 23 with “less severe acute tubular lesions.”
Acute Obstetric Complications
Since 1971, when DeLee
117 reported a state of “temporary hemophilia” in patients with premature separation of the placenta and a macerated dead fetus, it has been evident that a consumptive thrombohemorrhagic state is observed in a wide range of obstetric complications, including
αbruptio placentae, retained dead fetus, amniotic fluid embolism, saline-induced or septic
abortion, and occasionally toxemia. The site of consumption may be limited to a single extravascular locale (the uterus), disseminated in the blood, or present in both locations; the pathologic mechanism may involve coagulation, fibrinolysis, or both simultaneously; and the broadest range of tempo may occur, from the fulminant onset of an
abruptio placentae to the chronic, low-grade effects of the retained dead fetus syndrome.
Pregnancy is associated with a baseline hypercoagulable state characterized by increased levels of procoagulant factors, low protein S levels, and reduced fibrinolytic activity.
118,119 Pregnancy predisposes patients to DIC for at least four reasons: (a) a hypercoagulable state, manifested by low-grade thrombin generation, with elevated levels of fibrin monomer complexes and fibrinopeptide A; (b) reduced fibrinolytic activity because of increased plasma levels of PAI-1; (c) decline in the plasma free protein S; and (d) during labor, leakage of TF from placental tissue into the maternal circulation. DIC may be difficult to diagnose during pregnancy because of the high initial levels of coagulation factors such as fibrinogen, factor VIII, and factor VII,
120,121 but progressive reductions in these factors can help confirm or exclude the diagnosis. Thrombocytopenia may be particularly helpful in determining whether DIC is present, provided other causes are excluded.
122
Abruptio Placentae
Placental abruption is a leading cause of perinatal death.
123 Older multiparous women or patients with one of the hypertensive disorders of pregnancy are at highest risk. The severe hemostatic failure accompanying abruptio placentae is the result of acute DIC following the introduction of large amounts of TF into the circulation from the damaged placenta and uterus.
124 Amniotic fluid activates coagulation
in vitro, and the degree of placental separation correlates with the extent of DIC, suggesting that leakage of thromboplastin-like material from the placental system is responsible for DIC. Abruptio placentae occurs in 0.2% to 0.4% of pregnancies, but only 10% of these cases are associated with DIC.
119,122 Although in the classic study by Pritchard and Brekken
119 only 38% of patients had hypofibrinogenemia (<150 mg/dL), most patients had a relatively low fibrinogen for that stage of pregnancy. Rapid volume replacement and evacuation of the uterus is the treatment of choice.
123 Transfusion of cryoprecipitate, fresh-frozen plasma, and platelets should be given for profuse bleeding, but blood components may not be necessary in less severe cases, as coagulation factors increase rapidly following delivery. Heparin or antifibrinolytic agents are not indicated.
Amniotic Fluid Embolism
Amniotic fluid embolism was firmly established as a syndrome by the clinical and histopathologic changes described by Steiner and Lushbaugh
125 in 1941. Its incidence is 1:8,000 to 1:80,000 live births and is responsible for 10% of maternal deaths in the United States.
126 Patients predisposed to amniotic fluid embolism are multiparous women whose pregnancies are postmature and women undergoing a tumultuous labor. Amniotic fluid is introduced into the maternal circulation through tears in the chorioamniotic membranes, rupture of the uterus, and injury of uterine veins.
126 The trigger of DIC probably is TF present in amniotic fluid.
127,128 The mechanical obstruction of pulmonary blood vessels by fetal debris, meconium, particulate matter in the amniotic fluid enhances local fibrin-platelet thrombus formation and fibrinolysis. The extensive occlusion of the pulmonary arteries and an acute anaphylactoid response reminiscent of severe systemic inflammatory response syndrome provoke sudden dyspnea, cyanosis, acute cor pulmonale, left-ventricular dysfunction, shock, and convulsions. In patients who survive the initial episode, excessive bleeding follows in 40% of cases, usually after a latent period of 0.5 to 4 hours, and the great majority have laboratory evidence of consumption.
126 In the National Registry for amniotic fluid embolism (46 patients), cardiac arrest occurred in 87% and mortality was 61%, but only 15% of patients survived neurologically intact.
129 Seventy-nine percent of fetuses survived, but only 50% were neurologically unimpaired.
Hemorrhage is particularly severe from the atonic uterus, puncture sites, and GI tract. The best prospect for decreasing mortality lies in early termination of parturition in patients at high risk and prevention of hypertonic and tetanic uterine contractions during labor. When the syndrome is recognized, immediate termination of pregnancy under pulmonary and cardiovascular support is essential. Vigorous supportive therapy directed toward gas exchange with mechanical respiratory assistance and fluid and blood replacement to treat severe hypotension are essential. In patients in whom continued entry of amniotic fluid debris maybe contributing to further intravascular clotting, heparin is a reasonable adjunct to therapy.
121 When bleeding is the major manifestation, heparin may increase bleeding, especially if adequate replacement therapy for consumed clotting factors has not been achieved. In addition to replacement therapy for treatment of dangerous bleeding, fibrinolytic inhibitors may be helpful, but the same precautions regarding their use apply as in other settings of DIC.
Septic Abortion
In contrast to the relatively mild clinical state of saline-induced abortion, septic abortion heralds the most fulminant type of severe DIC. In a group of more than 6,000 patients who had undergone abortion, 16% were infected, 4% of whom had shock and DIC, leading to death in one-half of these seriously affected patients.
130 Any of a large number of bacterial organisms, both aerobic and anaerobic, may be the inciting agent; a combination of severe hypotension and DIC complicates the clinical picture. Once established, the clinical symptoms, laboratory manifestations, and principles of therapy are as for any severe infection leading to shock, with the added consideration that evacuation of the uterus and sometimes hysterectomy are required to eliminate the infection. These patients can be the most refractory to therapy, and vigorous attention must be paid to proper antibiotic choice, maintenance of blood volume and electrolyte balance, and replacement of deficient hemostatic factors. The use of heparin in these circumstances is controversial, and there is little evidence of its benefit. As with other circumstances of acute, severe DIC, fibrinolytic inhibitors should be withheld so as not to impair the physiologic fibrinolytic response in the microcirculation of ischemic organs.
Acute Fatty Liver
Acute fatty liver of pregnancy is a rare disorder that occurs during the third trimester of pregnancy,
131 which can lead to hepatic failure, encephalopathy, and maternal and fetal death.
132,133,134,135 In 15% to 20% of cases, acute fatty liver of pregnancy is associated with fetal homozygosity or compound heterozygosity
for long-chain acyl-CoA dehydrogenase (LCAD) deficiency,
136 one mutation (G1528C) accounting for 65% to 90% of cases. The precise mechanism by which LCAD deficiency in the fetus causes the severe liver disease in the heterozygous mother is unclear. The typical histologic feature of acute fatty liver disease of pregnancy is microvesicular fatty infiltration of the liver, which is characterized by severe liver dysfunction, renal failure, hypertension, and DIC.
133,137 Exceedingly low levels of AT and other laboratory signs of DIC were observed in a series of 28 patients, but no definite clinical benefit from AT concentrate infusion was achieved.
137 The primary therapy for these patients is early delivery and supportive care, which yield a maternal survival of 90% and perinatal survival of more than 85%
133,138 Pancreatitis is a potentially lethal complication of acute fatty liver of pregnancy.
139
Preeclampsia and “Hemolysis, Elevated Liver Enzymes, and a Low Platelet Count” (HELLP Syndrome)
The existence of DIC as a constant feature in patients with preeclampsia or eclampsia is controversial.
19,140,141 A critical analysis concluded that the thrombocytopenia in preeclamptic patients stems from endothelial injury rather than DIC,
140 but, other investigators provide evidence for DIC,
142,143 including a good correlation between the clinical severity and abnormalities in platelet counts and fibrin(ogen) degradation products.
141 The syndrome of hemolysis (H), elevated liver enzymes (EL), low platelet count (LP), and severe epigastric pain is a complication of pregnancy-induced hypertension.
144 Seventy percent of the cases occur during the third trimester of pregnancy and 30% occur during the postpartum period.
145 Hemolysis, elevated liver enzymes, and a low platelet count (HELLP) syndrome occurs more often in whites, multipara, and women older than 35 years.
146 Liver biopsy shows fibrin deposition, and laboratory tests are consistent with DIC in a significant proportion of patients.
145,147,148 Hepatic imaging in 33 patients revealed subcapsular hematomas in 13 and intraparenchymal hemorrhage in 6.
149 Although the majority of patients do not have laboratory evidence of DIC, a significant proportion have a low fibrinogen level (<300 mg/dL), which frequently is associated with evidence of abruptio placentae. MOD manifested by acute renal failure, ascites, pulmonary edema, and severe hemorrhage resulting from DIC may develop, leading to significant maternal and perinatal mortality. Management of patients with HELLP syndrome consists of supportive care, careful monitoring, and blood component replacement therapy.
150 With few exceptions, immediate delivery, not necessarily by Caesarian section, is indicated. HELLP syndrome tends to recur in subsequent gestations.
151
Liver Disease
Advanced liver disease may result in chronic low-grade DIC (see below). However, complications of acute or chronic liver disease may trigger episodes of an acute coagulopathy, most notably in patients with peritoneovenous (LeVeen or Denver) shunts or fatty liver of pregnancy. As reviewed by LeVeen et al.,
152 peritoneovenous shunting has a mortality of <1% in “uncomplicated cirrhosis,” and postoperative coagulopathies can largely be avoided. However, a substantial number of patients develop DIC shortly after shunt placement. Ragni et al.
153 detected DIC in 10 of 11 such patients, and Rubinstein et al.
154 found DIC after 14 of 27 shunt procedures and a greater tendency for this complication in patients with cirrhosis.
Acute Hemolysis and Transfusion Reactions
Acute hemolysis such as that after the transfusion of 500 mL or more of incompatible blood (major mismatch) can produce a hemorrhagic diathesis characterized by laboratory changes of hypofibrinogenemia and thrombocytopenia.
2 This pattern is compatible with acute DIC, with variable effects of intravascular thrombi on renal function depending on the acuteness and degree of defibrination and the additional presence of hypovolemia or hypotension. However, massive hemolysis (other than that associated with a major mismatch) does not uniformly produce DIC, as Mannucci et al.
155 observed in 28 patients with glucose-6-phosphate dehydrogenase deficiency who were exposed to fava beans. The DIC secondary to massive hemolysis is self-limiting, and in the absence of serious end-organ damage, patients improve rapidly with supportive measures. Treatment depends on maintenance of blood volume and organ perfusion and on blood replacement with compatible erythrocytes. DIC that accompanies incompatible blood transfusion may result from extensive antigen-antibody reaction causing release of elastase and TNF-α from neutrophils and activation of monocytes that express TF and complement, leading to assembly of the membrane attack complex inflicting damage to endothelial cells.
156,157
 The Field of Hemostasis and Thrombosis: Selected Translational Achievements
The Field of Hemostasis and Thrombosis: Selected Translational Achievements
 Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
Structure and Function of Vitamin K-Dependent Coagulant and Anticoagulant Proteins
 Integrín αIIbβ3 and Platelet Aggregation
Integrín αIIbβ3 and Platelet Aggregation
 Inherited Thrombocytopenias
Inherited Thrombocytopenias
 Unusual Sites of Arterial Occlusion
Unusual Sites of Arterial Occlusion
 Blood Management in the Cardiovascular Surgical Patient
Blood Management in the Cardiovascular Surgical Patient



