Once considered exceptionally rare, congenital thrombocytopenias are increasingly recognized as a heterogeneous group of disorders characterized by a reduction in platelet number and a bleeding tendency that may range from very mild to life threatening. Although some of these disorders affect only megakaryocytes and platelets, others involve different cell types and may result in characteristic phenotypic abnormalities. This review elaborates the clinical presentation and laboratory manifestations of common congenital thrombocytopenias in addition to exploring our understanding of the molecular basis of these disorders and therapeutic interventions available.
Key points
- •
Congenital thrombocytopenias are a heterogeneous group of disorders characterized by a reduction in platelet counts that may or may not be apparent at birth.
- •
Congenital thrombocytopenias are often accompanied by platelet function abnormalities—depending on the specific defect.
- •
Bleeding manifestations vary significantly and range from mild to life threatening.
- •
Certain congenital thrombocytopenias, such as MYH9-related disease (MYH9-RD), Wiskott-Aldrich syndrome (WAS), and thrombocytopenia–absent radius (TAR) syndrome, may be associated with classic phenotypic features that aid in their diagnosis.
- •
Advances in molecular genetics have greatly improved understanding of the underlying pathogenesis.
- •
Management depends on the severity of bleeding and presence of associated phenotypic manifestations.
Introduction
Congenital thrombocytopenias are a rare, heterogeneous group of disorders that result in early-onset thrombocytopenia with marked variability in bleeding manifestations ranging from mild to severe, life-threatening bleeds. Although some cases remain unclassifiable, recent advances in molecular genetics have dramatically improved understanding of the underlying pathogenesis of several of these conditions. Given the heterogeneity of these syndromes, there are several methods of classification. They may be classified according to the underlying genetic mutation ( Table 1 ), size of platelets ( Table 2 ), or associated abnormalities of platelet function. In this review, the syndromes have been grouped by the mode of inheritance as autosomal dominant, autosomal recessive, and X-linked.
| Syndrome (OMIM Entry) | Inheritance Pattern | Gene Involved | Chromosomal Location | Associated Findings |
|---|---|---|---|---|
| MYH9-RD (multiple) | Autosomal dominant | MYH9 | 22q12–13 | Döhle-like leukocyte inclusion bodies, nephritis, sensorineural hearing loss, and presenile cataracts |
| Paris-Trousseau (Jacobsen) syndrome (188025; 147791) | Autosomal dominant | FLI1 | 11q23.3–24 | Cognitive and facial abnormalities |
| PT-VWD (177820) | Autosomal dominant | GP1BA | 17p13 | Low VWF ristocetin cofactor assay, decrease in HMWVW multimers, increased low-dose RIPA; needs to be differentiated from type IIb VWD. |
| Radioulnar synostosis with amegakaryocytic thrombocytopenia (605432) | Autosomal dominant | HOXA11 | 17p15.2 | Radioulnar synostosis, clinodactyly, syndactyly, hip dysplasia, sensorineural hearing loss, and progression to pancytopenia |
| FPD/AML (601399) | Autosomal dominant | RUNX1 | 22q22 | Platelet aggregations shows impaired aggregation to collagen and epinephrine; 35% of the cohort may go on to develop AML. |
| ANKRD26 -related thrombocytopenia (nd) | Autosomal dominant | ANKRD26 | 10p2 | Normal-sized platelets with bone marrow shows dysmegakaryopoiesis; possible increased risk of leukemia |
| CYCS-related thrombocytopenia (612004) | Autosomal dominant | CYCS | 7p15.3 | None |
| TUBB1-related macrothrombocytopenia (nd) | Autosomal dominant | TUBB1 | 6p21.3 | None |
| BSS (231200) | Autosomal recessive | GP1BA, GP1BB, GP9 | 17p13, 22q11, 3q21 | Platelet aggregation studies show absent ristocetin-induced response. |
| GPS (139090) | Autosomal recessive (mostly) | NBEAL2 | 3p21 | None |
| CAMT (604498) | Autosomal recessive | MPL | 1p34 | Progression to pancytopenia |
| TAR syndrome (274000) | Autosomal recessive | RBM8A | 1q21.1 | Bilateral absent radii with thumb present; lower limb, cardiac, gastrointestinal, and renal anomalies |
| Thrombocytopenia associated with sitosterolemia (210250) | Autosomal recessive | ABCG5, ABCG8 | 2p21 | Stomatocytosis, tendon xanthomas, and premature atherosclerosis |
| WAS (XLT) (301000) | X-linked | WAS | Xp11.22 | Recurrent infections, eczema, with a risk of autoimmunity and lymphoid malignancy |
| XLT with dyserythropoiesis (300367; 314050) | X-linked | GATA1 | Xp11.23 | Dyserythropoietic anemia |
| FLNA-related thrombocytopenia (nd) | X-linked | FLNA | Xq28 | Possible association with periventricular nodular heterotopia |
| Small Platelets (MPV <7 fL) | Normal-Sized Platelets (MPV 7–11 fL) | Large Platelets (MPV >11 fL) |
|---|---|---|
| WAS XLT | CAMT TAR Radioulnar synostosis with amegakaryocytic thrombocytopenia FPD/AML ANKRD26 -related thrombocytopenia CYCS -related thrombocytopenia | MYH9-RD BSS GPS VCFS XLT with dyserythropoiesis PT-VWD Paris-Trousseau (Jacobsen) syndrome TUBB1 -related macrothrombocytopenia Thrombocytopenia associated with sitosterolemia |
There are various circumstances under which a patient may be worked up for congenital thrombocytopenias—these include a history of bleeding or easy bruising, family history of thrombocytopenia, or an incidental finding of thrombocytopenia detected on blood count done for an unrelated indication. Given that several congenital thrombocytopenias are associated with only a mild reduction of platelet counts and no hemorrhagic symptoms, a major concern is misdiagnosis of these conditions as the more common immune thrombocytopenia (ITP) and subsequent exposure of patients to unnecessary and often inappropriate therapies, such as splenectomy.
The first step in the work-up of such patients is a thorough bleeding history and family history, which may help differentiate acquired and congenital defects as well as ascertain the inheritance pattern of the bleeding disorder. In the past decade, significant work has been done to develop standardized bleeding questionnaires, which can be used in both adult and pediatric populations. Subsequent steps involve performing a complete blood cell count and examination of a peripheral smear. Pseudothrombocytopenia, a common phenomenon caused by platelet clumping in the presence of ethylenediaminetetraacetic acid (EDTA), accounts for 15% to 20% of all cases of isolated thrombocytopenias and is easily ruled out with evaluation of a peripheral smear. The initial inspection of blood smear also helps determine platelet size (small, normal, or large), detect the appearance of agranular platelets, visualize leukocyte inclusions, or detect other red and white cell abnormalities. Further characterization involves bone marrow aspiration and biopsy as well as platelet aggregation studies. Given the emerging data showing a genotype-phenotype correlation for several congenital thrombocytopenias, efforts should be made to refer samples to specialized laboratories that can perform such testing. The purpose of this review is to provide an updated approach to the clinical presentation, laboratory investigation, and management of congenital thrombocytopenias.
Introduction
Congenital thrombocytopenias are a rare, heterogeneous group of disorders that result in early-onset thrombocytopenia with marked variability in bleeding manifestations ranging from mild to severe, life-threatening bleeds. Although some cases remain unclassifiable, recent advances in molecular genetics have dramatically improved understanding of the underlying pathogenesis of several of these conditions. Given the heterogeneity of these syndromes, there are several methods of classification. They may be classified according to the underlying genetic mutation ( Table 1 ), size of platelets ( Table 2 ), or associated abnormalities of platelet function. In this review, the syndromes have been grouped by the mode of inheritance as autosomal dominant, autosomal recessive, and X-linked.
| Syndrome (OMIM Entry) | Inheritance Pattern | Gene Involved | Chromosomal Location | Associated Findings |
|---|---|---|---|---|
| MYH9-RD (multiple) | Autosomal dominant | MYH9 | 22q12–13 | Döhle-like leukocyte inclusion bodies, nephritis, sensorineural hearing loss, and presenile cataracts |
| Paris-Trousseau (Jacobsen) syndrome (188025; 147791) | Autosomal dominant | FLI1 | 11q23.3–24 | Cognitive and facial abnormalities |
| PT-VWD (177820) | Autosomal dominant | GP1BA | 17p13 | Low VWF ristocetin cofactor assay, decrease in HMWVW multimers, increased low-dose RIPA; needs to be differentiated from type IIb VWD. |
| Radioulnar synostosis with amegakaryocytic thrombocytopenia (605432) | Autosomal dominant | HOXA11 | 17p15.2 | Radioulnar synostosis, clinodactyly, syndactyly, hip dysplasia, sensorineural hearing loss, and progression to pancytopenia |
| FPD/AML (601399) | Autosomal dominant | RUNX1 | 22q22 | Platelet aggregations shows impaired aggregation to collagen and epinephrine; 35% of the cohort may go on to develop AML. |
| ANKRD26 -related thrombocytopenia (nd) | Autosomal dominant | ANKRD26 | 10p2 | Normal-sized platelets with bone marrow shows dysmegakaryopoiesis; possible increased risk of leukemia |
| CYCS-related thrombocytopenia (612004) | Autosomal dominant | CYCS | 7p15.3 | None |
| TUBB1-related macrothrombocytopenia (nd) | Autosomal dominant | TUBB1 | 6p21.3 | None |
| BSS (231200) | Autosomal recessive | GP1BA, GP1BB, GP9 | 17p13, 22q11, 3q21 | Platelet aggregation studies show absent ristocetin-induced response. |
| GPS (139090) | Autosomal recessive (mostly) | NBEAL2 | 3p21 | None |
| CAMT (604498) | Autosomal recessive | MPL | 1p34 | Progression to pancytopenia |
| TAR syndrome (274000) | Autosomal recessive | RBM8A | 1q21.1 | Bilateral absent radii with thumb present; lower limb, cardiac, gastrointestinal, and renal anomalies |
| Thrombocytopenia associated with sitosterolemia (210250) | Autosomal recessive | ABCG5, ABCG8 | 2p21 | Stomatocytosis, tendon xanthomas, and premature atherosclerosis |
| WAS (XLT) (301000) | X-linked | WAS | Xp11.22 | Recurrent infections, eczema, with a risk of autoimmunity and lymphoid malignancy |
| XLT with dyserythropoiesis (300367; 314050) | X-linked | GATA1 | Xp11.23 | Dyserythropoietic anemia |
| FLNA-related thrombocytopenia (nd) | X-linked | FLNA | Xq28 | Possible association with periventricular nodular heterotopia |
| Small Platelets (MPV <7 fL) | Normal-Sized Platelets (MPV 7–11 fL) | Large Platelets (MPV >11 fL) |
|---|---|---|
| WAS XLT | CAMT TAR Radioulnar synostosis with amegakaryocytic thrombocytopenia FPD/AML ANKRD26 -related thrombocytopenia CYCS -related thrombocytopenia | MYH9-RD BSS GPS VCFS XLT with dyserythropoiesis PT-VWD Paris-Trousseau (Jacobsen) syndrome TUBB1 -related macrothrombocytopenia Thrombocytopenia associated with sitosterolemia |
There are various circumstances under which a patient may be worked up for congenital thrombocytopenias—these include a history of bleeding or easy bruising, family history of thrombocytopenia, or an incidental finding of thrombocytopenia detected on blood count done for an unrelated indication. Given that several congenital thrombocytopenias are associated with only a mild reduction of platelet counts and no hemorrhagic symptoms, a major concern is misdiagnosis of these conditions as the more common immune thrombocytopenia (ITP) and subsequent exposure of patients to unnecessary and often inappropriate therapies, such as splenectomy.
The first step in the work-up of such patients is a thorough bleeding history and family history, which may help differentiate acquired and congenital defects as well as ascertain the inheritance pattern of the bleeding disorder. In the past decade, significant work has been done to develop standardized bleeding questionnaires, which can be used in both adult and pediatric populations. Subsequent steps involve performing a complete blood cell count and examination of a peripheral smear. Pseudothrombocytopenia, a common phenomenon caused by platelet clumping in the presence of ethylenediaminetetraacetic acid (EDTA), accounts for 15% to 20% of all cases of isolated thrombocytopenias and is easily ruled out with evaluation of a peripheral smear. The initial inspection of blood smear also helps determine platelet size (small, normal, or large), detect the appearance of agranular platelets, visualize leukocyte inclusions, or detect other red and white cell abnormalities. Further characterization involves bone marrow aspiration and biopsy as well as platelet aggregation studies. Given the emerging data showing a genotype-phenotype correlation for several congenital thrombocytopenias, efforts should be made to refer samples to specialized laboratories that can perform such testing. The purpose of this review is to provide an updated approach to the clinical presentation, laboratory investigation, and management of congenital thrombocytopenias.
Autosomal dominant thrombocytopenias
MYH9-Related Disease
Introduction
MYH9-RD is an autosomal dominant condition presenting with macrothrombocytopenia and leukocyte inclusion bodies (Döhle-like bodies) present since birth and with a risk of developing nephropathy, sensorineural deafness, and presenile cataracts later in life, secondary to heterozygous mutations in the MYH9 gene encoding for the nonmuscle myosin heavy chain IIA (NMMHC-IIA).
In 1909, Richard May first described the presence of Döhle-like inclusion bodies in the leukocytes of subjects with giant platelets. Subsequently, in 1945, Robert Hegglin described a family in which macrothrombocytopenia and leukocyte inclusion bodies were transmitted in an autosomal dominant fashion. This led to the term, May-Hegglin anomaly , used to describe the triad of thrombocytopenia, giant platelets, and leukocyte inclusion bodies. Over the next 50 years, 3 distinct syndrome complexes, namely Epstein syndrome, Fechtner syndrome, and Sebastian syndrome, described the association of nephritis, sensorineural deafness, and presenile cataracts with dominantly inherited thrombocytopenia and leukocyte inclusion bodies. It was only in the year 2000 that it was shown by 2 independent groups that all these syndromes derive from mutations in the MYH9 gene, and the term, MYH9-RD, is now preferred.
Molecular defect
More than 40 mutations have been identified in MYH9 gene located on chromosome 22q12–13. They mostly consist of heterozygous missense mutations, suggesting that the pathology is either a dominant negative effect (influencing the function of the normal protein) or haploinsufficiency (inadequate amount of protein). NMMHC-IIA, a 453-kDA protein, is a hetero-hexatetramer that contains 2 heavy chains and 2 pair of light chains, which form a dimer through the interactions between the α-helical tail domains. Each heavy chain contains an N-terminal globular head (exons 1–18), which has ATPase and actin-binding activity, a neck region (exon 19), and a C-terminal α-helical coiled-coil tail domain. NMMHC-IIA plays an important role in several cellular processes requiring force and translocation, such as motility, cytokinesis, phagocytosis, vesicular trafficking, maintenance of stress fibers, and focal adhesions. In addition, 2 recent studies have shown that myosin IIA also has a key role as a negative regulator of platelet formation by inhibition of proplatelet extension via the Rho/ROCK pathway. In vitro studies have subsequently shown that the MYH9 mutation is associated with complete loss of proplatelet extension of megakaryocytes, and it is hypothesized that patients with MYH9-RD have ectopic migration of proplatelets and premature release of platelets into the bone marrow niches, resulting in thrombocytopenia.
Clinical presentation and laboratory evaluation
Bleeding tendency in patients with MYH9-RD tends to be mild to moderate with epistaxis, easy bruising, and menorrhagia being the commonly reported symptoms, though life-threatening bleeds, including intracranial hemorrhage, have also been described. It is not unusual, however, for asymptomatic adults to be diagnosed with MYH9-RD after routine blood work. In such situations, an important concern is misdiagnosis of such patients as having ITP and subsequent exposure to inappropriate therapies, including intravenous immunoglobulin (IVIG), steroids, and splenectomy. An absence of family history of macrothrombocytopenia does not rule out MYH9-RD because 35% of the mutations may be sporadic.
Pecci and colleagues, in a seminal study of 108 consecutive patients with gene sequencing–proven MYH9-RD, estimated the prevalence of nephritis, sensorineural hearing loss, and presenile cataracts to be 28%, 60%, and 16%, respectively. Mean age of onset of nephritis and presenile cataracts was 23 (±9) years and 23 (±6) years, respectively, whereas there was homogenous distribution of age of onset for sensorineural hearing loss among all ages. In the same study, the genotype-phenotype correlation of MYH9-RD was elaborated. Mutations in the N-terminal head domain were more likely associated with nephritis ( P <.001), sensorineural hearing loss ( P <.001), and more severe thrombocytopenia ( P = .002) compared with mutations in the tail domain, which usually resulted in hematological manifestations alone.
Mean platelet counts are typically approximately 20 × 10 9 /L to 130 × 10 9 /L with an elevated mean platelet volume (MPV). Automated cell counters can mistake these giant platelets as erythrocytes and may report spuriously low platelet counts, emphasizing the importance of visualizing the blood film. In the study by Pecci and colleagues, mean platelet count of the 108 patients, as measured by automated counters, was 31 × 10 9 /L; however, the 60 patients who had manual cell counts had a significantly higher mean platelet count at 68 × 10 9 /L. Specific diagnostic criteria have been suggested, which combine MPV and mean platelet diameter (MPD) to help distinguish congenital thrombocytopenias from ITP. In a prospective study of 15 patients with MYH9-RD, 20 with Bernard-Soulier syndrome (BSS) and 50 with ITP, MPV, and MPD were significantly higher in patients with congenital thrombocytopenias. Receiver operating characteristic curves identified that an MPV greater than 12.4 fL had 83% sensitivity and 89% specificity whereas an MPD greater than 3.3 μm had 89% sensitivity and 88% specificity in differentiating congenital thrombocytopenia from ITP. Combining the MPV and MPD resulted in sensitivity and specificity of 97% and 89%, respectively.
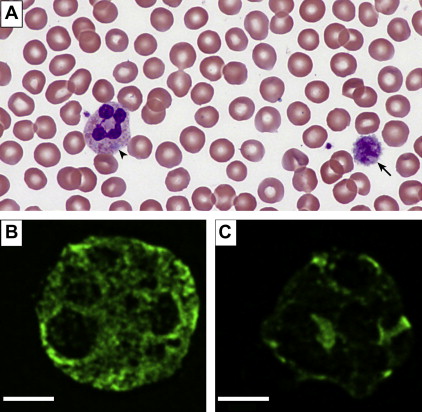
Döhle-like inclusion bodies in leukocytes can be appreciated on Wright-Geisma–stained blood films in approximately 40% to 80% of patients. These light blue–appearing inclusions are aggregates of NMMHC-IIA observed in the cytoplasm of some neutrophils ( Fig. 1 A ). However, since these inclusions are not always present, immunofluorescence staining of blood films using anti–NMMHC-IIA antibody is more sensitive and is able to detect smaller and abnormally localized aggregates (see Fig. 1 B, C). In a prospective study of a 118 consecutive subjects evaluated for MYH9-RD, using genetic analysis as the gold standard, the sensitivity and specificity of this technique were estimated at 100% and 95%, respectively. Given increasing evidence of genotype-phenotype correlation and its impact on screening for renal disease and hearing loss, whenever possible, gene sequencing of the MYH9 gene should be pursued. Given the clustered distribution of mutations, Balduini and colleagues have suggested a tiered approach with the hot exons, namely, 1, 16, 26, 30, 38, and 40, being sequenced first followed by exons 10, 24, 25, 31, and 37, and, finally, the remaining codons. Patients with confirmed MYH9-RD should be referred to specialized facilities for ongoing screening for nephritis, hearing loss, and cataracts.
Paris-Trousseau (Jacobsen) Syndrome
Paris-Trousseau (Jacobsen) syndrome is an autosomal dominant macrothrombocytopenia first described in 1993 in a woman and her child. It is characterized by moderate thrombocytopenia with a subpopulation of platelets demonstrating giant alpha granules (1–2 μm diameter) that stain red on Wright-Giemsa stain ( Fig. 2 ). Bone marrow evaluation shows marked dysmegakaryopoiesis with an increase in number of small immature megakaryocytes. Paris-Trousseau platelets, when stimulated by thrombin, fail to release their alpha granule contents. This syndrome is associated with constitutional deletions of the distal portion of chromosome 11 (11q23.3–24), including the friend leukemia integration ( FLI1 ) locus. FLI1, a member of the ETS group of transcription factors, regulates the expression of several genes involved in megakaryocyte differentiation and maturation, including ITGA2 , GP9 , GPIBA , and c-MPL, and its disruption in mice is associated with a dramatic decrease in megakaryocyte number and size. Furthermore, overexpression of FLI1 in CD34 + cells from Paris-Trousseau syndrome patients was able to rescue megakaryopoiesis in vitro, thereby confirming this gene’s involvement in patient megakaryocyte and platelet abnormalities.
Favier and colleagues reported clinical, hematological, and molecular data on 10 pediatric patients with Paris-Trousseau syndrome. All patients had moderate thrombocytopenia at diagnosis (platelet counts 20–76 × 10 9 /L), although there was a trend toward increase in platelet counts a few years after initial diagnosis. Giant alpha granules were observed in 1% to 5% of the platelets, but they were not detected in bone marrow megakaryocytes or those grown in culture. They appeared with megakaryocyte maturation and were thought to result from fusion of alpha granules. Molecular analysis using fluorescence in situ hybridization demonstrated 11q23.3 deletion in all patients, and reverse transcription–polymerase chain reaction confirmed loss of 1 copy of the FLI1 gene in 9 of 10 patients. Clinically, individuals with Paris-Trousseau have a mild bleeding tendency, although the Jacobsen phenotype, which is thought to result from larger deletions on the long arm of chromosome 11, is more extensive with mental retardation, trigonocephaly, facial dysmorphism, and malformations of the heart, kidney, gastrointestinal tract, genitalia, and skeleton. The KIRREL3 gene has recently been implicated in the neurocognitive delay associated with Jacobsen syndrome.
Platelet-type von Willebrand Disease
Introduction
Platelet-type von Willebrand disease (PT-VWD) was first described by Weiss and colleagues in 1982, in 4 members of a family presenting with mild bleeding symptoms and intermittent thrombocytopenia. Similar to type 2B von Willebrand disease (VWD), the cohort had increased ristocetin-induced platelet aggregation (RIPA) and loss of high-molecular-weight von Willebrand (HMWVW) multimers. Unlike type 2B VWD, however, the primary defect was thought to exist in the platelets rather than the von Willebrand protein, and Weiss named the condition pseudo–von Willebrand disease . Since its initial description, 55 cases of PT-VWD from 23 families have been reported ( http://www.pt-vwd.org/ ).
PT-VWD is an autosomal dominant bleeding diathesis that occurs as a result of gain-of-function mutations in the GP1BA gene, coding for the platelet surface glycoprotein, GPIbα. This defect causes spontaneous binding of plasma von Willebrand factor (VWF) to platelets, with subsequent removal of the VWF-platelet complex from circulation, resulting in the thrombocytopenia and bleeding. By contrast, type 2B VWD occurs as a result of gain-of-function mutations in the A1 domain (exon 28) of the VWF gene. The phenotypic and laboratory manifestations of both PT-VWD and type 2B VWD are similar. However, given the distinct therapeutic modalities (platelet transfusion for PT-VWD; factor VIII [FVIII]/VWF concentrate for type 2B VWD), it is important to differentiate the 2 conditions.
Molecular defect
To date, 5 mutations causing PT-VWD have been described in the GP1BA gene. These include 4 missense mutations, resulting in single amino acid substitutions—Gly233Val, Met239Val, Gly233Ser, and Asp235Tyr —and a novel in-frame deletion of 27bp in the macroglycopeptide region. The amino acid residues, 233, 235, and 239, are situated within a β-hairpin loop in the crystalline structure of GP1bα, and it is hypothesized that these substitutions stabilize the loop, resulting in an increased affinity to VWF under nonshear conditions. A knock-in mouse model of PT-VWD with the Gly233Val mutation not only presents with a bleeding phenotype but also exhibits increase in splenic megakaryocytes, splenomegaly, and increased bone mass.
Clinical presentation and laboratory evaluation
Patients with PT-VWD typically present with mild to moderate mucocutaneous bleeding, which includes easy bruising, frequent epistaxis, menorrhagia, and postsurgical bleeding. Bleeding may become more severe under conditions of stress, pregnancy, and infection due to an increased release in the endogenous VWF under these conditions or after ingestion of aspirin and other drugs with antiplatelet activity.
Platelet counts may be normal, although intermittent mild to moderate thrombocytopenia may be appreciated in conditions of stress. Blood films often reveal large platelets (macrothrombocytes) with platelet clumping. FVIII and VWF antigen are typically normal to mildly reduced, with a discordant decrease in von Willebrand functional assays, such as VWF ristocetin cofactor assay and VWF collagen-binding assay, and reduction in HMWVW multimers. The most characteristic finding is enhanced RIPA at concentrations of 0.5 mg/mL or lower. The discrimination of PT-VWD from the more common type 2B VWD is difficult but clinically relevant. It is estimated that PT-VWD is underdiagnosed, with a recent international retrospective/prospective registry demonstrating that 15% of patients diagnosed with type 2B VWD actually had PT-VWD. The following tests may help differentiate the 2 conditions:
- 1.
Cryoprecipitate challenge: patient platelets (platelet-rich plasma or washed platelets) are challenged with cryoprecipitate or a commercial VWF concentrate. Only PT-VWD (and not type 2B VWD) platelets demonstrate aggregation on addition of a concentrated form of VWF. False-positive results have been reported with this technique.
- 2.
RIPA mixing assay: patient platelets (platelet-rich plasma or washed platelets) are mixed with normal plasma, and aggregation is measured in an aggregometer at low-dose (0.5 mg/mL) ristocetin concentration. Platelets from PT-VWD (not type 2B VWD) aggregate under these conditions.
- 3.
Flow cytometry assay: the increased affinity of VWF to platelets induced by ristocetin was recently demonstrated using flow cytometry. Using mixing tests, the investigators were able to differentiate PT-VWD from type 2B VWD.
- 4.
Genetic analysis: gene sequencing of the GP1BA gene or the A1 region (exon 28) of the VWF gene with identification of mutation remains the gold standard for diagnosis of PT-VWD or type 2B VWD, respectively. This test is only available in specialized centers.
Radioulnar Synostosis with Amegakaryocytic Thrombocytopenia
The association between amegakaryocytic thrombocytopenia and radioulnar synostosis was first reported in 2 unrelated, nonconsanguinous families by Thompson and colleagues. Both fathers and 4 affected children presented with proximal fusion of the radius and ulna, resulting in limited pronation and supination of the forearm. Additional phenotypic features included clinodactyly, syndactyly, hip dysplasia, and sensorineural hearing loss. Although the fathers had no hematological manifestations, all 4 children had thrombocytopenia at birth. Initial bone marrow evaluations showed normal cellularity with decreased megakaryocytes, although 3 children eventually progressed to trilineage pancytopenia within 6 months to 6 years, with follow-up bone marrow examinations showing generalized hypocellularity. The fourth child had normalization of platelet counts on follow-up.
Genetic studies identified a single base pair deletion on exon 2 of the HOXA11 gene, resulting in a translational frameshift followed by a premature stop codon, which was thought to result in the truncation of this transcription factor within its DNA-binding domain. These homeobox ( HOX ) genes encode DNA-binding proteins that regulate gene expression central to bone morphogenesis and hematopoietic cell differentiation. Subsequently, more cases of amegakaryocytic thrombocytopenia in association with radioulnar synostosis have been reported, although HOXA11 mutations have not been consistently identified. The autosomal dominant inheritance, mild deformity of the forearm, and persistent thrombocytopenia progressing to pancytopenia differentiate this condition from TAR syndrome (discussed later).
Familial Platelet Disorder with Propensity for Myeloid Malignancy
The first pedigree of familial platelet disorder with a propensity for myeloid malignancy (FPD/AML) was described by Dowton and colleagues in 1985, in 22 members of a kindred with autosomal dominant thrombocytopenia, 6 of whom went on to develop hematological malignancies. In 1996, Ho and colleagues reported genetic linkage to chromosome 21q22, and, in 1999, the same group identified heterozygous mutations in RUNX1 located on 21q22. FPD/AML is currently recognized as a rare autosomal dominant disorder characterized by qualitative and quantitative platelet defects, with a predisposition to develop myeloid malignancies later in life, secondary to heterozygous germline mutations in the RUNX1 gene . FPD/AML is rare with approximately 30 pedigrees reported in the literature.
The RUNX1 gene spans 260 kilobases (kb) of genomic sequence on chromosome 21, contains 12 exons, and exhibits a complex pattern of regulated expression at the level of transcription. RUNX1, also known as core binding factor A2, complexes with core binding factor beta (CBFB) to form a heterodimeric, core binding transcription factor that regulates many genes important for hematopoiesis. A highly conserved DNA-binding region called the RUNT homology domain, located near the N-terminus of RUNX1, both enables the dimerization with CBFB and directs binding of RUNX1 to DNA sequences of target genes. Mutations in the RUNT homology domain are most commonly reported in kindreds with FPD/AML, although large deletions and mutations in the C-terminus are also described.
The clinical presentation of FPD/AML is variable, with most patients presenting with mucocutaneous bleeding symptoms, including easy bruising, epistaxis, and bleeding with dental and minor surgery. Thrombocytopenia is mild (100–150 × 10 9 /L) and is associated with normal-sized platelets. The platelet function analyzer-100 (PFA-100) shows prolonged closure times with both collagen-ADP and collagen-epinephrine cartridges, and platelet aggregation typically shows impaired aggregation with collagen and epinephrine. Electron microscopy findings in older cohorts have reported dense granule deficiency. The incidence of acute myeloid leukemia and/or myelodysplastic syndrome in patients with FPD/AML is estimated at 35%, with a median age of diagnosis of 33 years.
Autosomal recessive thrombocytopenias
Bernard-Soulier Syndrome
Introduction
In 1948, 2 French hematologists, Jean Bernard and Jean Pierre Soulier, described a young man from a consanguineous family presenting with severe bleeding symptoms, thrombocytopenia, and large platelets. BSS is now recognized as an autosomal recessive macrothrombocytopenia secondary to quantitative deficiency or qualitative defects in the platelet GPIb-IX-V complex, resulting in decreased platelet adhesion to the subendothelium. BSS is rare, with an estimated prevalence of less than 1 in 1,000,000, although this number is thought an underestimation secondary to misdiagnosis and under-reporting. Heterozygous carriers for BSS mutations are usually asymptomatic, although a proportion of them may have mild bleeding symptoms (discussed later).
Molecular defect
The central function of the GPIb-IX-V complex is to ensure normal primary hemostasis by mediating platelet adhesion to the subendothelium at sites of vascular injury, through binding with VWF. GPIb-IX-V is composed of 4 transmembrane polypeptide subunits, synthesized by 4 separate genes: GPIbα ( GP1BA ), GPIbβ ( GP1BB ), GPIX ( GP9 ), and GPV ( GP5). The products of these genes assemble on the maturing megakaryocyte in the bone marrow to form the GPIb-IX-V complex, which is stabilized via filamin A. Mutation of the GP1BA gene is associated most frequently with BSS, although mutations in GP1BB and GP9 (but not GP5 ) also are described. More than 50 distinct mutations have been identified that affect the biosynthesis and/or trafficking of the GPIb-IX-V complex through the Golgi apparatus and endoplasmic reticulum of the megakaryocytes. No correlation between the type of mutation and bleeding phenotypes is observed.
Murine models have shown that the bleeding defect in BSS is predominantly due to loss of the adhesive function of GPIbα of the GPIb-IX-V complex. GPIbα contains the VWF-binding site and 2 thrombin-binding sites. In addition, it also binds to P-selectin, thrombospondin-1, FVIIa, FXI, FXII, αMβ2, and high-molecular-weight kininogen. Thus, abnormal bleeding in BSS is thought to result from impairment of GPIbα-VWF interaction at the site of vessel wall damage, although VWF-mediated high shear, platelet-platelet interaction has also been implicated. Additionally, abnormal membrane development during megakaryocyte maturation and impaired proplatelet formation may also contribute to the phenotype.
Clinical presentation and laboratory evaluation
Although there is marked heterogeneity in the severity and frequency of bleeding between individuals, most patients with BSS develop significant mucocutaneous hemorrhage in early childhood. Common bleeding manifestations include petechiae, epistaxis, gingival bleeding, menorrhagia, and hemorrhage with trauma or surgery. Angiodysplasia in patients with BSS resulting is severe gastrointestinal bleeding has been described. Often, bleeding and bruising are more significant, as expected from the degree of thrombocytopenia.
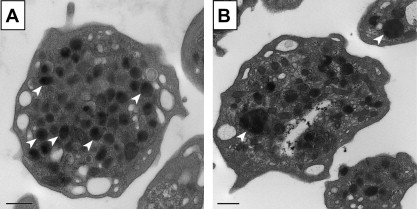
Characteristic laboratory findings include moderate thrombocytopenia, giant platelets ( Fig. 3 ), and absent RIPA. Platelet counts range from low (<30 × 10 9 /L) to normal, with presence of a small number of giant platelets with a rounded shape (MPV 11–16 fL; diameter 4–10 μm). Automated cell counters can mistake these giant platelets for lymphocytes and may report spuriously low platelet counts, thereby emphasizing the importance of visualizing the blood film. Similar to MYH9-RD, major concerns are misdiagnosis of patients having ITP and subsequent exposure to inappropriate therapies, including splenectomy (discussed earlier). Platelet aggregation studies classically show deficient RIPA, which does not correct with addition of normal plasma, as is the case in VWD. PFA-100 reveals a prolonged closure time (>300 seconds) with both the collagen-ADP and collagen-epinephrine cartridges. Flow cytometry analysis, using a panel of specific monoclonal antibodies (anti–CD42a-d) measuring components of the GPIb-IX-V complex, confirms the diagnosis. Electron microscopy, although not necessary for diagnosis, shows increased cytoplasmic vacuoles and membrane complexes in giant platelets and megakaryocytes.