Abstract
Congenital adrenal hyperplasia (CAH) refers to a group of autosomal recessive disorders in which genetic enzyme deficiencies impair normal steroid synthesis. The production of cortisol in the zona fasciculata of the adrenal cortex occurs in five major enzyme-mediated steps, and deficiency in any one of these leads to CAH. The most common form is 21-hydroxylase deficiency (21-OHD), which accounts for over 90% of CAH cases. Impaired cortisol synthesis, the common feature to all forms of CAH, leads to chronic elevation of adrenocorticotropic hormone (ACTH) and overstimulation of the adrenal cortex resulting in adrenal hyperplasia. Impaired enzyme function at each step of adrenal cortisol biosynthesis leads to an increase in precursors and deficient products. This chapter will focus on CAH owing to 21-OHD, the most frequent form of CAH.
Keywords
congenital adrenal hyperplasia, salt-wasting (SW), simple-virilizing (SV), 21-hydroxylase deficiency (21-OHD), phenotype–genotype correlation, virilization
Introduction
Background, Incidence, Prevalence
Congenital adrenal hyperplasia (CAH) refers to a group of autosomal recessive disorders in which genetic enzyme deficiencies impair normal steroid synthesis. The production of cortisol in the zona fasciculata of the adrenal cortex occurs in five major enzyme-mediated steps, and deficiency in any one of these leads to CAH. The most common form is 21-hydroxylase deficiency (21-OHD), which accounts for over 90% of CAH cases. Impaired cortisol synthesis, the common feature to all forms of CAH, leads to chronic elevation of adrenocorticotropic hormone (ACTH) and overstimulation of the adrenal cortex, resulting in adrenal hyperplasia. Impaired enzyme function at each step of adrenal cortisol biosynthesis leads to an increase in precursors and deficient products. The clinical and hormonal findings of five forms of CAH are summarized in Table 14.1 . This chapter will focus on CAH owing to 21-OHD, the most frequent form of CAH.
| Enzyme deficiency | Substrate | Product | Androgen | Mineralocorticoid |
|---|---|---|---|---|
| Steroidogenic acute regulatory protein | Cholesterol | Mediates cholesterol transport across mitochondrial membrane | Deficiency * | Deficiency ** |
| 3β-dehydrogenase | Pregnenolone, 17-OH pregnenolone, DHEA | Progesterone, 17-OHP, 4-androstenedione | Deficiency * | Deficiency ** |
| 17α-hydroxylase | Pregnenolone, progesterone | 17-OH pregnenolone, 17-OH progesterone (17-OHP) | Deficiency * | Excess † |
| 21-hydroxylase | Progesterone, 17-OH progesterone | Deoxycorticosterone (DOC), 11-deoxycortisol | Excess ‡ | Deficiency ** |
| 11β-hydroxylase | Deoxycorticosterone | Corticosterone | Excess ‡ | Excess † |
* Males are undervirilized at birth.
** Associated with salt wasting.
† Associated with hypertension.
Data from nearly 6.5 million newborn screens worldwide for 21-hydroxylase deficiency indicate that classical CAH occurs in 1:13,000 to 1:15,000 live births. Nonclassical 21-OHD CAH (NC21-OHD) is more common. The incidence of NC21-OHD in the heterogeneous population of New York City is one in 100, making NC21-OHD one of the most frequent recessive disorders in humans. NC21-OHD is particularly frequent in Ashkenazi Jews, in whom one in three are carriers of the allele, and one in 27 are affected with NC21-OHD. Steroid 11 deficiency (11-OHD) is the second most common cause of CAH, accounting for 5–8% of all cases. It occurs in one in 10,000 live births in the general population and is more common in some populations of North African origin. In Moroccan Jews, for example, the disease incidence of 11-OHD was initially estimated to be one in 5000 live births; subsequently, it was shown to occur less frequently, but remains more common in Moroccan Jews than in other populations. 17α-Hydroxylase deficiency is found to be common in Mennonite descendants of Dutch Frieslanders and the Brazilian population. The disease incidence of congenital lipoid adrenal hyperplasia is shown to be common in Japan and Korea. The other forms are considered rare diseases, and the incidence is unknown in the general population.
A very rare form of CAH not included in this table is cytochrome P450 oxidoreductase deficiency. It is characterized by partial, combined deficiencies of P450C17 (17α-hydroxylase), P450C21 (21-hydroxylase), and aromatase. Affected girls are born with ambiguous genitalia, indicating intrauterine androgen excess. However, virilization does not progress after birth. The 17-hydroxyprogesterone levels are elevated, as in 21-hydroxylase deficiency, while androgen levels are low; cortisol may be normal but is poorly responsive to ACTH. Conversely, affected boys may be born undervirilized, owing to 17α-hydroxylase deficiency. Boys and girls can also present with bone malformations, resembling a pattern seen in patients with Antley–Bixler syndrome.
Steroid 21 Hydroxylase Deficiency (21-OHD)
CAH owing to 21-OHD is divided into classical and nonclassical forms. Classical CAH is further divided into salt-wasting (SW) and simple-virilizing (SV) forms. The SW form is the most severe form of CAH and is characterized by inadequate aldosterone synthesis, which places patients at risk for SW crises, whereas the SV form is distinguished by the ability to conserve salt. Both the SW and SV forms result in genital ambiguity in the female. Females affected with classical forms of CAH are born with genital ambiguity termed “46XX, disorder of sex development.” In 21-OHD, the precursors to the 21-hydroxylase enzyme are shunted into the androgen pathway. Virilization of the female genitalia begins in utero as a result of fetal adrenal androgen production. Males do not exhibit genital ambiguity because the major source of androgens in males is the testes, not the adrenals. The nonclassical form of CAH causes postnatal symptoms of hyperandrogenism that may present any time after birth, including adulthood. Table 14.2 summarizes the main clinical characteristics of all forms of 21-OHD CAH.
| Feature | Salt-wasting (classical) | Simple-virilizing (classical) | Nonclassical |
|---|---|---|---|
| Prenatal genital virilization | Present in females | Present in females | Absent |
| Postnatal hyperandrogenic signs | Males and females | Males and females | Variable presentation |
| Salt-wasting | Present | None | Absent |
| Cortisol deficiency | Present | Present | Absent |
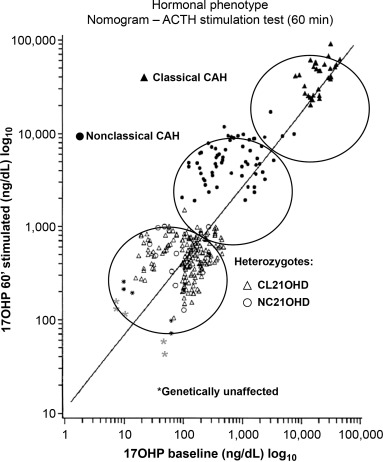
In all three forms of 21-OHD CAH, both affected males and females experience the effects of postnatal hyperandrogenism unless treatment intervenes. These hyperandrogenic symptoms include premature development of pubic and axillary hair, frontal hair loss, severe acne, advanced somatic and epiphyseal maturation and induced central precocious puberty (a condition in which puberty develops unusually early or rapidly). Early epiphyseal maturation may lead to compromised final adult height. Milder symptoms and/or later presentation are typical in nonclassical 21-OHD CAH owing to genetic mutations associated with less severe enzymatic defects. A synthetic ACTH stimulation test can differentiate 21-OHD from other forms of CAH. A logarithmic nomogram was developed to provide hormonal standards for diagnosis and further assignment of the 21-OHD type by relating baseline to ACTH-stimulated serum concentrations of 17-OHP (see Fig. 14.1 ).
Genetic pathophysiology
Hormonally and clinically defined forms of 21-OHD CAH are associated with distinct genotypes characterized by varying enzyme activity, demonstrated through in vitro expression studies. The gene encoding 21-hydroxylase is a microsomal cytochrome P450 termed cytochrome P450, family 21, subfamily A, polypeptide 21 ( CYP21A2 ; previously called P450c21B , CYP21B , or CYP21 ) (Online Mendelian Inheritance in Man [OMIM] database number 201910) mapped to the short arm of chromosome 6, at locus 6p21 within the human leukocyte antigen (HLA) complex (see Fig. 14.2 ). CYP21A2 and its homolog, the pseudogene CYP21A1P , alternate with two genes, C4B and C4A , that encode the two isoforms of the fourth component of the serum complement system.
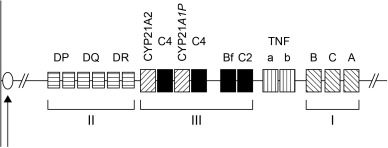
More than 140 mutations have been described, including point mutations, small deletions, small insertions, and complex rearrangements of the gene. About 20% of the mutant alleles are meiotic recombinations deleting a 30-kb gene segment that encompasses the 3′ end of the CYP21A1P pseudogene, all of the adjacent C4B complement gene, and the 5′ end of CYP21A2 , producing a nonfunctional chimera. Gene conversion causes mutations through transfer of deleterious mutations from the pseudogene to the active gene. De novo mutations in the active gene can also occur, but account for a small percentage of mutations in CYP21A2 . These mutations result in complete or partial insufficiency of 21-hydroxylase activity based on in vitro expression studies. The common mutations in CYP21A2 are summarized in Table 14.3 . Approximately 95–98% of the mutations causing 21-OHD have been identified through molecular genetic studies of gene rearrangement and point mutations arrays. Some mutations are particularly prevalent in certain ethnic groups, indicating a high ethnic specificity.
| Exon/intron | Mutation type | Mutation | Phenotype | Severity of enzyme defect (% enzyme activity) | References |
|---|---|---|---|---|---|
| |||||
| Exon 1 | Missense mutation | P30L | NC | Mild (30–60%) | Tusie-Luna, 1991 |
| Exon 7 | Missense mutation | V281L | NC | Mild (20–50%) | Speiser, 1988 |
| Exon 8 | Missense mutation | R339H | NC | Mild (20–50%) | Helmberg, 1992 |
| Exon 10 | Missense mutation | P453S | NC | Mild (20–50%) | Helmberg, 1992; Owerbach, 1992 |
| |||||
| Deletion | 30 kb deletion | – | SW | Severe (0%) | White, 1984 |
| Intron 2 | Aberrant splicing of intron 2 | 656 A/C-G | SW, SV | Severe (ND) | Higashi, 1988 |
| Exon 3 | Eight-base deletion | G110 8nt | SW | Severe (0%) | White, 1994 |
| Exon 4 | Missense mutation | I172N | SV | Severe (1%) | Amor, 1988; Tusie-Luna, 1990 |
| Exon 6 | Cluster mutations | I236N, V237E, M239K | SW | Severe (0%) | Amor, 1988; Tusie-Luna, 1990 |
| Exon 8 | Nonsense mutation | Q318X | SW | Severe (0%) | Globerman, 1988 |
| Exon 8 | Missense mutation | R356W | SW, SV | Severe (0%) | Chiou, 1990 |
| Exon 10 | Missense mutation | R483P * | SW | Severe (1–2%) | Wedell, 1993 |
Related posts:
 Developmental Abnormalities of the Thyroid
Genetics of Hyperparathyroidism Including Parathyroid Cancer
Developmental Abnormalities of the Thyroid
Genetics of Hyperparathyroidism Including Parathyroid Cancer
 Vitamin D Disorders
Genetic Considerations in the Evaluation of Menstrual Cycle Irregularities
Vitamin D Disorders
Genetic Considerations in the Evaluation of Menstrual Cycle Irregularities
 Androgen Insensitivity Due to Mutations of the Androgen Receptor
Androgen Insensitivity Due to Mutations of the Androgen Receptor
 Multiple Endocrine Neoplasia Type 1 (MEN1)
Multiple Endocrine Neoplasia Type 1 (MEN1)
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


