chromosomal gains and losses, phenomena that favor amplification of oncogenes and loss of tumor suppressors.10 Chromosomal segregation defects may account for this background of chromosomal instability (CIN), as illustrated by the segregation factor Bub1 in mice,11 but few specific gene defects are implicated with confidence. The remaining fraction of CRCs appears euploid at the level of whole chromosomes but may carry thousands of point mutations, small deletions, and insertions near nucleotide repeat tracts, a defect known as microsatellite instability (MSI).12 Molecular determinants of progression in MSI+ adenomas differ from those associated with CIN; for example, BRAF V600E mutations occur more commonly in MSI+ serrated adenomas than in other subtypes.13 Hypermutability with CIN or MSI results in many inconsequential or detrimental “passenger” mutations, an important consideration that focuses attention on “driver” changes. Such changes are distinguished by their appearance in a significant proportion of tumor specimens and, ideally, by laboratory demonstration of their contribution toward malignancy.
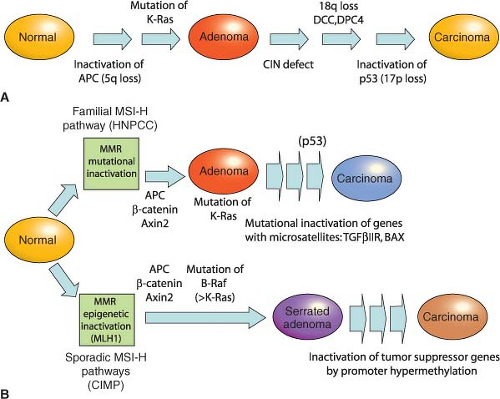 FIGURE 19.1 Genetic pathways to colorectal carcinoma. All colorectal cancers (CRCs) arise within benign adenomatous precursors, fueled by mutations that serially enhance malignant behavior. Mutations that activate the Wnt signaling pathway seem to be necessary initiating events, following which two possible courses contribute to accumulation of additional mutations. A: Chromosomal instability is a feature of up to 80% of CRCs and is commonly associated with activating KRAS point mutations and loss of regions that encompass P53 and other tumor suppressors on 18q and 17p, often but not necessarily in that order. B: About 20% of CRCs are euploid but defective in DNA mismatch repair (MMR), resulting in high microsatellite instability (MSI-Hi). MMR defects may develop sporadically, associated with CpG island methylation (CIMP), or as a result of familial predisposition in hereditary nonpolyposis colorectal cancer (HNPCC). Mutations accumulate in the KRAS or BRAF oncogenes, p53 tumor suppressor, and in microsatellite-containing genes vulnerable to MMR defects, such as TGFβIIR. Epigenetic inactivation of the MMR gene MLH1 and activating BRAF point mutations are especially common in serrated adenomas, which progress in part through silencing of tumor suppressor genes by promoter hypermethylation. Progression from adenoma to CRC takes years to decades, a process that accelerates in the presence of MMR defects. |
adenomas.15 Reduced pericentromeric methylation might decrease the fidelity of chromosomal segregation, and altered methylation and loss of imprinting at the IGF2 locus are associated with increased CRC risk,16 suggesting broad effects of global hypomethylation on cell growth. However, because some animal models show increased tumor susceptibility with global hypomethylation,17 whereas ApcMin mice that lack or overexpress the de novo DNA methyltransferase DNMT3B show reduced or increased progression of small adenomas, respectively,18,19 its precise significance is unclear. Against the background of genomewide hypomethylation, a subset of CRCs show coordinate hypermethylation of characteristic CpG-rich promoter islands, conferring the CpG island methylator phenotype (CIMP), with transcriptional attenuation of associated genes, including tumor suppressors such as HIC1 and the secreted Wnt-inhibiting secreted Frizzled-related proteins (sFRPs).20,21 Adenomatous precursors of CIMP cancers show the distinctive histology of sessile serrated adenomas, with dysplasia within an architectural pattern typical of hyperplastic polyps.
TABLE 19.1 GENETICS OF INHERITED COLORECTAL TUMOR SYNDROMES | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ||||||||||||||||||||||||||||||||||
degradation. Thus, inactivating APC or activating CTNNB1 mutations, two alternative lesions in CRC, have the same effect: constitutive, Wnt-independent stabilization of β-catenin (Fig. 19.2). Accumulated β-catenin translocates to the cell nucleus, where it acts as a transcriptional coactivator for the T-cell factor/lymphoid enhancer factor (TCF/LEF) family of transcription factors. Nuclear β-catenin provides TCF/LEF proteins with an activating partner, resulting in transcription of target genes.38 Of the four known TCF/LEF proteins, TCF4 is the most important in normal bowel epithelium and CRC.34,39 Among the many components of the Wnt signaling cascade, rare AXIN2 mutations of uncertain significance are reported in MSI+ cases,40 but mutations in CRC are otherwise found only in APC and CTNNB1.
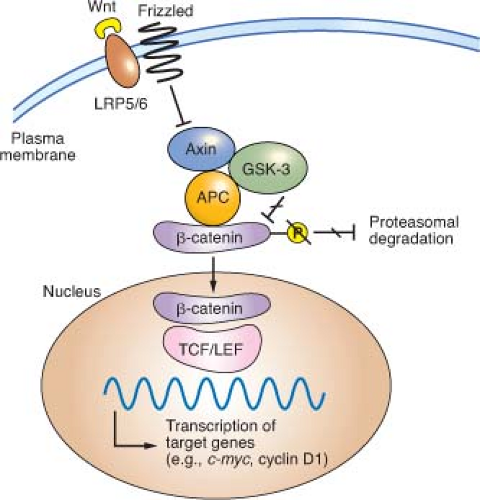 FIGURE 19.2 Outline of Wnt signaling, the key driver pathway in colorectal cancer. Members of the Wnt family of glycoprotein morphogens bind the cell surface coreceptors Frizzled and LRP5/6. In the absence of Wnt binding, normal cells use a complex containing adenomatous polyposis coli (APC), Axin, and other cytoplasmic proteins to promote glycogen synthase kinase (GSK)-3β-mediated phosphorylation of the β-catenin N-terminus, which targets β-catenin for proteasomal degradation (from ref. 37.). Binding of a Wnt ligand to Frizzled and its obligate coreceptor LRP5/6 antagonizes the APC/Axin destruction complex, stabilizing β-catenin (CTNNB1), which moves into the nucleus and coactivates genes through T-cell factor/lymphoid enhancer factor (TCF/LEF) transcription factors. Either of the two principal gatekeeper events in colorectal cancer, inactivating APC or activating CTNNB1 mutations, results in constitutive, Wnt-independent stabilization of β-catenin and unregulated activation of the cognate transcriptional program. Wnt signaling in the intestine is normally confined to crypt progenitors, and its aberrant activation by APC or CTNNB1 mutations confers a permanent cryptlike state that favors cell replication. |
TABLE 19.2 CRITERIA FOR CLINICAL DIAGNOSIS OF HEREDITARY NONPOLYPOSIS COLORECTAL CANCER | |||||||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| |||||||||||||||||||||||||||||||||||||||
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


