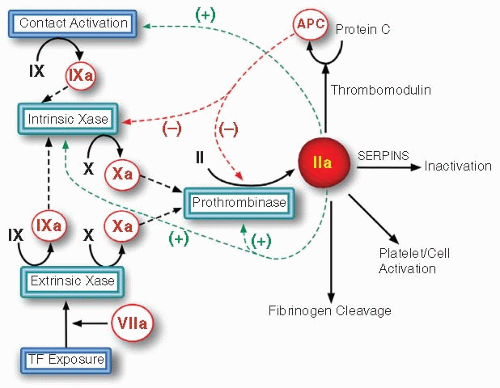
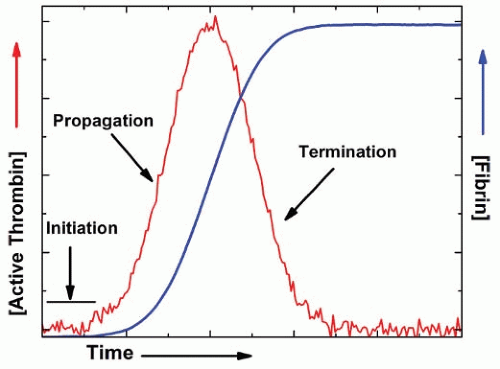
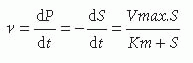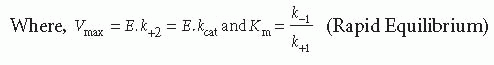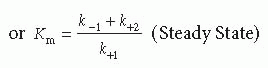Blood coagulation is initiated by the exposure of tissue factor to flowing blood following vascular damage.1,2 A series of highly specific proteolytic activation steps ensue to convert an inactive circulating precursor to an active product. The product in turn participates in a subsequent activation reaction (FIGURE 4.1). Thrombin, formed as a result of these reactions, is the key effector product of coagulation as it is the proteinase responsible for the cleavage of fibrinogen to form the insoluble fibrin meshwork. It also activates platelets, other blood cells, and the endothelium by cleavage of the protease-activated receptors. Thrombin is a self-limiting product. Early in the coagulation response, it functions as a positive feedback effector. It greatly increases flux through the cascade by activating cofactors and factor XI necessary for the earlier steps of coagulation. Later in the response, it acts as a negative feedback effector by binding thrombomodulin (TM) and catalyzing protein C activation, which in turn proteolytically inactivates V and VIII. Their inactivation reduces flux toward thrombin formation. Active thrombin and the other proteinases of coagulation are eliminated by reacting with circulating serine proteinase inhibitors. As a result of this central regulatory role played by thrombin and the critical role played by proteinase inhibitors, the fate of active thrombin formed following the initiation of coagulation exhibits a transient profile, in which the profile for fibrin formation is dictated by the amplitude and timing of the peak as well as its integrated area, reflecting the total amount of active thrombin produced (FIGURE 4.2).
THE PROBLEM
Regulated enzymic function plays a dominant role in modulating the hemostatic response to vascular damage. Proteinase specificity reflects the ability of any one enzyme to selectively act on its cognate substrate in the complex milieu of blood. This becomes an especially important aspect of coagulation enzymology because the coagulation proteinases are trypsin-like enzymes belonging to the S1 family of subclan PA(S) of serine endopeptidases.3 Their catalytic domains are highly structurally homologous to each other and to the digestive enzymes chymotrypsin and trypsin, the archetypic members of the clan.3 The nature of the problem is highlighted by the fact that while trypsin can, in theory, act to cleave after any lysine and arginine in any protein, the homologous coagulation proteinases act with marked specificity to cleave after just one or two arginines in a limited subset of structurally related proteins. Thus, rigidly limited proteinase specificity lies at the heart of the hemostatic response. Flowing blood also constrains coagulation enzyme function to a physical regime that differs markedly from those encountered in the “test tube” where formalisms deal with circumstances in which substrates can be quantitatively converted to product. Instead, normal hemostasis derives from activation reactions that occur at a reactive patch, representing the area of vascular damage, to which substrates are constantly being delivered by flowing blood and products are washed away. Membrane-dependent enzyme function localizes many of the coagulation reactions to the site of vascular damage to which inert precursors are delivered by flowing blood. Products that escape the reactive patch are rapidly inactivated downstream by proteinase inhibitors. This chapter focuses on the unique strategies employed by the enzymes of blood coagulation to achieve these essential aspects of function.
PROTEINASE FORMATION
As defined in subsequent chapters, specialized structural domains present in the NH2-terminal half of each of these zymogens contribute to a variety of aspects of zymogen and resulting proteinase function, including membrane binding by the vitamin K-dependent zymogens. However, the zymogens all possess a (chymo)trypsinogen-like domain at their respective COOH-termini that is converted to the catalytic domain by proteolytic activation.4,5 Similarity between the catalytic domains of the different enzymes has been established by an abundance of high-resolution x-ray structures. Based on this identity, Bode et al.6 introduced a uniform strategy of numbering residues in the catalytic domain or its precursor. Topologically equivalent and conserved residues within the family are numbered using the sequence numbers for the equivalent residues in chymotrypsinogen (e.g., Ic16, chymotrypsinogen numbering denoted by the subscripted c). When insertion loops unique to the individual coagulation proteins are encountered, they are denoted alphabetically, resuming with the equivalent numbering at the end of the insertion (e.g., Pc204-Fc204A-Nc204B-Nc205). This provides a uniform and facile approach for considering the structural correlates of catalytic function in this family of enzymes.
Proteinase formation results from cleavage of the peptide bond following Rc15, regardless of other cleavages that may occur in the processing of any particular zymogen by its cognate enzyme. This cleavage reaction produces a new N-terminus with variations on the sequence Ic16-Vc17-Gc18-Gc19 seen in trypsin. The nascent sequence inserts in a sequence-specific manner into an N-terminal binding cleft to form a key salt bridge with Dc194. The term “Molecular Sexuality,” coined by Bode, provides a graphic description of the sequence-specific nature of the insertion process.7,8 Proteinase formation is impaired in mutants bearing an altered nascent sequence in spite of cleavage following Rc15.9,10 Conversely, trypsinogen can be conformationally activated even without cleavage by the addition of a I-V dipeptide but not V-I.7 Ion pairing is associated with changes in four activation domains (Table 4.1) that are flanked by highly conserved glycines, referred to as glycine hinges.11 Changes in these activation domains about the glycine hinges lead to optimization of the primary specificity pocket for substrate binding and a flip in the Gc193 amide bond to form the oxyanion hole necessary for catalysis. These transitions instill proteolytic activity in the product by enhancing the ability of the active site to engage the substrate and by increasing catalytic power. Depending on proteinase, changes in other regions important for ligand binding may also result.
FIGURE 4.1 The blood coagulation cascade. The scheme illustrates zymogen activation steps in the pathway for thrombin formation. The positive feedback reactions of thrombin are illustrated by the green arrows and the negative feedback reactions that result from the action of activated protein C are shown by red arrows.
FIGURE 4.2 Expected fate of active thrombin following initiation of coagulation. The regulated hemostatic response results in the transient formation of active thrombin. Positive feedback reactions of thrombin produced at low levels in the initiation phase lead to amplified thrombin generation during the propagation phase. Negative regulation by activated protein C and inactivation by serpins predominate late in the response leading to depletion of active thrombin. The expected course of fibrin formation is also illustrated.
Relatively subtle differences in the main chain traced in x-ray structures of the zymogen and the corresponding proteinase yield the impression that the zymogen to proteinase transition is accompanied by subtle changes in protein structure. However, the effect of cleavage and salt bridge formation is primarily reflected in the ordering of the activation domains, which are otherwise disordered in the zymogen.12 Thus, very significant ordering effects result from proteinase formation and an associated 104-105-fold increase in catalytic activity. Since maturation of the proteinase is rooted in conformational transitions that follow irreversible cleavage of the zymogen, it seems reasonable that formation of the fully stabilized proteinase is irreversibly favored following cleavage. However, this idea is not consistent with observations made in early studies of chymotrypsin13 and studies of VIIa,14,15 and is increasingly questioned in the case of thrombin.12,16,17 Conversely, while the uncleaved zymogen is prevented from making the conformational transitions required for proteinase formation, evidence from the related proteins of fibrinolysis suggests that the uncleaved protein can be imbued with catalytic activity as a result of contributions of alternate side chains in the vicinity of Dc194.18 Thus, although the relationship between cleavage following Rc15 and proteinase formation is tacitly assumed to be absolute, it need not be so. Ligand-dependent effects on the various transitions along the pathway of conversion of zymogen to proteinase may represent an underappreciated but prevalent strategy for enzyme regulation in hemostasis.
Table 4.1 Activation domains of zymogen activation
Domain
Functional Correlate
c16-c21
N-terminal insertion region
c142-c152
Autolysis loop
c184-c194
Region encompassing bottom of specificity pocket, oxyanion hole, and ion pair partner.
c216-c223
Residues framing entrance to S1 site
CATALYTIC SPECIFICITY
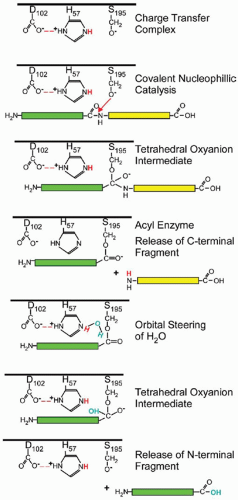
Specificity for peptide bond hydrolysis results from a catalytic triad of side chains provided by Hc57, Sc195 and Dc102 that are precisely oriented at the active site.19 A charge transfer complex is formed by the abstraction of a proton from Sc195 by Hc57, which is in turn stabilized by long-range electrostatic interactions with Dc102 (FIGURE 4.3). Precise orientation of the scissile bond in the substrate allows covalent nucleophillic catalysis at the carbonyl group by electronegative Sc195 to yield a tetrahedral oxyanion intermediate. The tetrahedral intermediate is stabilized by electrostatic interactions with the oxyanion hole. Transfer of a proton to the tetrahedral adduct by the action of Hc57 as a general acid catalyst releases the COOH-terminal fragment, while the NH2-terminal fragment acylates Sc195 and remains covalently tethered to the enzyme. Water diffusing into the active site is oriented through orbital steering by Hc57, which then acts as a general base catalyst to abstract a proton allowing for the hydrolysis of the acyl-enzyme intermediate. The resulting tetrahedral intermediate is also stabilized by the oxyanion hole before it collapses to release the NH2-terminal fragment, returning the catalytic triad to its initial state for the next round of catalysis. These steps outline the range of strategies in chemical catalysis employed by serine proteinases to catalyze peptide bond hydrolysis.
BINDING SPECIFICITY
Peptide bond cleavage requires the precise positioning of the polypeptide substrate to subtend the carbonyl group of the scissile bond to the catalytic residues within the active site cleft. Binding specificity that arises from such positioning drives the ability of proteinases within this structurally homologous family to act with high selectivity on their cognate protein substrates. This is accomplished by structures on the proteinase complementary to structural features on the substrate. Selectivity of the coagulation proteinases for cleavage following arginine indicates that they are trypsin-like with a binding pocket optimized to accommodate this side chain leading to the presentation of the scissile bond to the catalytic residues. Because the coagulation enzymes do not cleave after all arginines, additional residues must also contribute to substrate-binding specificity. Drawing from studies with the less specific proteinases in this family, concepts are grounded in the idea that two to four residues preceding the scissile bond are major determinants of binding specificity with their side chains engaging complementary sites in the enzyme. The terminology of Schechter and Berger20 is followed, where substrate side chains are numbered S1, S2, S3, and so on extending N-terminal to the scissile bond and S1′, S2′, S3′, and so on extending in the C-terminal direction. Sites on the enzyme complementary to these substrate side chains sites and flanking the catalytic site are labeled P1, P2, P3 or P1′, P2′, P3′ and so on. Ideas regarding the determinants of substrate specificity have historically been developed from work done with peptidyl substrates with readily measureable leaving groups (FIGURE 4.4). As a result, and because of high homology with trypsin, the principal focus has been on the role played by the S residues and P sites in determining substrate specificity with less emphasis on the S′ residues and P′ sites.
FIGURE 4.3 Peptide bond hydrolysis catalyzed by serine proteinases.
KINETICS OF ENZYME ACTION
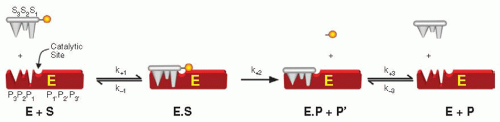
Cleavage of a typical peptidyl substrate by a coagulation proteinase requires binding of the substrate to complementary sites on the enzyme to form a Michaelis complex (ES). Positioning of the scissile bond at the catalytic triad leads to catalysis and release of the chromophore, followed by reversible peptide dissociation and a new round of catalysis. This represents a straightforward platform for the further consideration of the kinetics of enzyme action from simple measurements of the rate of product formation. Despite its simplicity, the scheme yields stiff differential equations that typically require single turnover studies on the millisecond timescale for the resolution of all stepwise rate constants.21 Although approaches have been presented to solve these rate constants from simple measurements of the rate of product formation at different viscosities, the extraordinary level of data precision required for reliable conclusions makes this approach impractical.22 Consequently, a meaningful quantitative analysis from simple measurements of the rate of substrate consumption and of product formation requires simplifying assumptions: (a) [E] « [S]. This is an appropriate and reasonable condition, given the catalytic power of the typical enzyme, (b) the initial velocity assumption requires that initial rates are measured before there is significant substrate consumption or product formation. This principally allows product inhibition effects to be ignored, although such effects need to be accounted for in the case of some tight-binding substrates/products, and (c) either the rapid equilibrium or the steady state assumption. In the former case, it is assumed that the binding and dissociation of substrate is very rapid relative to the rate constant for catalysis. In the latter, it is assumed that the system is in steady state during the rate measurement and distributions between the various enzyme species are invariant with time. This yields the familiar equation widely attributed to Michaelis and Menten,23 wherein the constants take on different meaning depending on the assumptions that apply:
FIGURE 4.4 Kinetic scheme for peptidyl substrate binding and cleavage.
E, S, and P refer to the total concentrations of enzyme, substrate, and product, k+2 (kcat) is the rate constant for catalysis, Vmax is the maximum rate of product formation at infinite S, and Km is the Michaelis constant. In the limiting case that ES and E are in rapid equilibrium, Km reflects the equilibrium dissociation constant (Kd) for substrate binding, but can be significantly larger than Kd in the steady-state case. Therefore, Km is a measure of the affinity of the enzyme for substrate with smaller values of Km reflecting tighter binding. At fixed E, the dependence of rate on S is defined by a rectangular hyperbola and Km is the concentration of S at which v = 0.5 Vmax. When acylation limits the rate constant for catalysis, the leaving group and peptidyl product are produced at an equal rate. When deacylation is rate limiting, the leaving group is produced more rapidly than the peptidyl product. Substrate species that display the latter properties are employed in burst kinetic reactions to determine the concentration of active proteinase.24
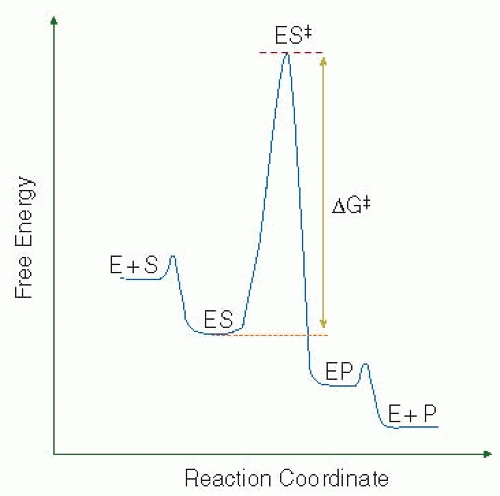
In the free-energy landscape for the reaction (FIGURE 4.5), the ES complex represents a ground state that needs to overcome the free-energy barrier imposed by the transition state before it can be converted to product. Thus, kcat is inversely proportional to the magnitude of the free-energy barrier (ΔG‡) separating the ground and transition states. Within this framework, an increased kcat requires that ΔG‡ be reduced by stabilizing the transition state and/or destabilizing the ground state. Both ground and transition state effects are accommodated in the catalytic efficiency term (kcat/Km), which represents the overall second order rate constant for the reaction. In the screening of different substrates with any particular enzyme, preference for a particular substrate is implied by a larger value for catalytic efficiency (also referred to as specificity constant). Considerations developed from peptidyl substrate cleavage studies continue to dominate ideas underlying substrate specificity, and analyses using different peptidyl substrates continue to represent a primary avenue for the investigation of the substrate preferences of the various coagulation proteinases.
FIGURE 4.5 Free energy diagram for peptidyl substrate hydrolysis by serine proteinases.
LIMITATIONS OF PEPTIDYL SUBSTRATE STUDIES
Oligopeptidyl substrates are microscopic in relation to the protein substrate and thereby interact in a limited way with the active site of the proteinase. While the protein substrates of the coagulation proteinases must bind to the active site before they are cleaved, these substrates approximate the size of the enzyme and could potentially engage the enzyme far more extensively, with such interactions contributing in a major way to binding specificity. Thus, while kinetic measurements with peptidyl substrates are facile and widely employed, they routinely fail to provide an explanation for cleavage site specificity within the protein substrate.25,26,27 This limitation is widely evident in the coagulation proteinases, where extended interactions between enzyme and substrate play a fundamental role in determining macromolecular substrate specificity.
PAN-SPECIFICITY OF THROMBIN
Thrombin is an atypical coagulation proteinase in that it cleaves numerous substrates in reactions that are essential for clot formation and its regulation. Although the S2-S1-S1′ sequence is P-R-S at several cleavage sites in its different substrates, a single consensus pattern flanking the S1 arginine fails to emerge. Early studies with thrombin revealed that proteolysis in its catalytic domain could greatly reduce its ability to cleave fibrinogen without affecting the rate of peptidyl substrate hydrolysis.28 Dissociation of the two functions provided an early indication that the basis for macromolecular substrate specificity of thrombin lies in enzymic structures beyond those relevant to the hydrolysis of peptidyl substrates. A series of elegant studies of the mechanism of inhibition of thrombin by hirudin from the laboratories of Stuart Stone and Jan Hofstengee established the central role of a basic region distant from the catalytic site in this high-affinity inhibition reaction.29,30,31,32,33 Subsequently, ample biochemical and structural evidence has established the central role played by this anion-binding exosite 1 (ABE1) region (FIGURE 4.6) in engaging acidic regions found in several thrombin substrates distant from the scissile bond. In these cases, substrate binding to ABE1 likely guides and facilitates active site engagement of structures surrounding the cleavage site leading to catalysis. The interaction with ABE1 can be a major determinant of binding affinity, as well as specificity, since proteolysis of the loops that constitute ABE1 or its occlusion by ligands can abrogate cleavage of substrates that engage thrombin in this way.28,34,35 These ideas provide a partial accounting for the strategies employed by thrombin to achieve pan-specificity without strict conservation of residues flanking cleavage sites in its various substrates.
ENZYME COMPLEXES OF COAGULATION
While thrombin acts on a range of protein substrates, the other proteinases of coagulation exhibit far more restricted substrate specificity. In addition, many of these reactions are catalyzed by enzyme complexes that assemble from membrane-dependent interactions between a proteinase and a cofactor protein. Thus, there is no basis, a priori, to presume that strategies utilized by thrombin for substrate recognition would apply to other reactions.
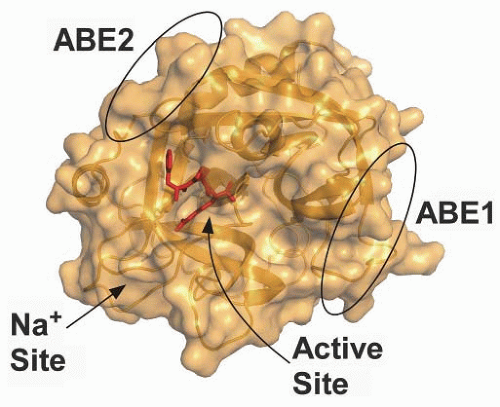
FIGURE 4.6 X-ray structure of thrombin. Structure of thrombin modified by D-FPR-chloromethyl ketone (1PPB) is shown as a rendered surface in the standard orientation.6 The active site is highlighted by the bound inhibitor shown as red sticks. The positions of anion-binding exosite 1 (ABE1) and exosite 2 (ABE2) are indicated as is the Na+ binding site.
The prothrombinase complex, the enzyme complex that converts the zymogen prothrombin to thrombin, is considered an archetype for the other complexes of coagulation. This is appropriate because of the wealth of biophysical and enzymologic information delineating function and the reversible binding interactions that lead to complex assembly and substrate recognition.1,4
Prothrombinase is composed of the serine proteinase, factor Xa, bound to factor Va, its protein cofactor, on a membrane surface (FIGURE 4.7). Although complex assembly in vivo requires exposing phosphatidylserine on the outer membrane of activated blood cells such as platelets, and possibly also the endothelium, most of the quantitative information in this system has been developed in defined systems with purified proteins and synthetic membranes containing phosphatidylcholine and phosphatidylserine (PCPS). While factor Xa is a fully competent serine proteinase, its ability to cleave prothrombin is minor in comparison to the rate observed following its incorporation into prothrombinase. Indeed, based on rates, the concentration of thrombin formed in 1 second by Xa saturably bound to Va and membranes would require approximately 5 days by the equivalent concentration of Xa in solution. Thus, the assembly of Xa into prothrombinase is required for rapid thrombin formation following initiation of coagulation, and it is prothrombinase rather than free Xa that is considered the physiologically relevant catalyst for rapid prothrombin activation following vascular damage. The other enzyme complexes are composed of homologous constituents that assemble in an analogous way with comparable functional consequences.
The functional consequences of the reversible interactions of Xa with membranes and Va are evident from steady-state kinetic constants for “prothrombin activation” determined using Xa in solution compared with Xa saturably bound to membranes or Xa saturated with Va and membranes (Table 4.2). Binding of Xa to membranes decreases the apparent affinity of the proteinase for prothrombin, while addition of saturating concentrations of cofactor does not affect Km further, but yields a large improvement in the rate constant for catalysis. At physiologic concentrations of prothrombin (1.4 µM), these kinetic constants reveal that the minimal activity of Xa in solution is increased approximately 500,000-fold upon its incorporation into prothrombinase. These types of observations are generalizable to the other complexes of coagulation. They also exhibit comparable changes in steady-state kinetic constants attributable to membrane binding and cofactor binding. Because these binding interactions are reversible, valid comparisons of the type shown here require knowledge of the bound concentrations of the proteinase in the various binary and ternary mixtures from individual binding constants established by independent physical measurement. Incomplete consideration of the degree of saturation of enzyme components can lead to incorrect conclusions and is probably the root of the persistent idea that prothrombinase assembled on platelets is a far superior catalyst to the complex assembled on synthetic membranes.36,37 Indeed, the reverse is probably true because of nonproductive interactions of enzyme components with the platelet surface.
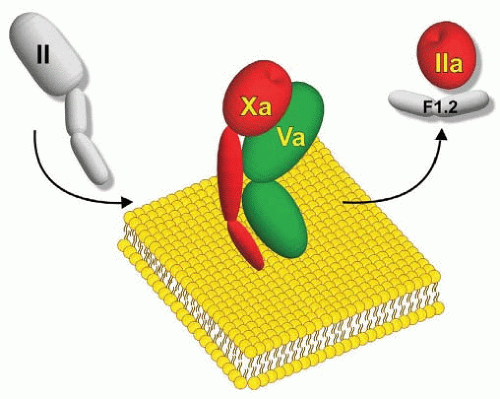
FIGURE 4.7 Schematic representation of prothrombinase. The complex assembles through membrane-dependent interactions of the proteinase Xa with the cofactor protein, factor Va, on membranes containing phosphatidyl-L-serine. The complex converts the zymogen, prothrombin (II) to thrombin (IIa) with the release of the N-terminal propiece (F12). Catalytically active species are colored red.
Table 4.2 Steady-state kinetic constants for prothrombin activationa
aKinetic constants for bovine prothrombin cleavage by bovine Xa alone,102 or for the activation of human prothrombin by human enzyme constituents in the presence of membranes.103
b The enzyme species correspond to Xa alone, Xa saturably bound to membranes (Xa/PCPS) and Xa assembled into prothrombinase (Xa/Va/PCPS) using saturating concentrations of membranes and Va.
c Relative rates at 1.4 µM prothrombin calculated from the steady-state kinetic constants.
Only gold members can continue reading. Log In or Register to continue