CLINICAL FEATURES
21-HYDROXYLASE (P450C21) DEFICIENCY
Patients with this commonly diagnosed enzyme deficiency cannot adequately synthesize cortisol. Insufficient cortisol synthesis results in overproduction of adrenal androgens, which are synthesized independently of 21-hydroxylase. Androgens induce somatic growth with inappropriately rapid advancement of linear growth, early epiphyseal fusion of the long bones, and short stature. Other features include precocious development of sexual hair, apocrine body odor, and penile or clitoral enlargement. Reduced fertility may be observed in both sexes. Clinical features are outlined in Table 77-1.
Females affected with the severe classic form of 21-hydroxylase deficiency are exposed to excess androgens pre-natally and are born with masculinized external genitalia (Fig. 77-2). If the disease goes undiagnosed and the infant is untreated, further virilization ensues (Fig. 77-3). Approximately 75% of patients with the classic form cannot synthesize aldosterone efficiently because of impaired 21-hydroxylation of progesterone; these salt-wasting individuals fail to conserve sodium normally and usually come to medical attention in the neonatal period with hyponatremia, hyperkalemia, and hypovolemic shock. These adrenal crises may prove fatal if proper medical care is not delivered. Patients with sufficient aldosterone production and no salt wasting who have signs of prenatal virilization and markedly increased production of hormonal precursors of 21-hydroxylase (e.g., 17-hydroxyprogesterone) are referred to as simple virilizers. Earlier confusion regarding the origins of these two classic phenotypes has been resolved to a large extent by the understanding of the molecular genetics of the disease, and allelic variation in the gene encoding active 21-hydroxylase (CYP21) appears to be responsible for most phenotypic variation, as is discussed later.
 FIGURE 77-2. External genitalia of a 2-month-old female infant with 21-hydroxylase deficiency. |
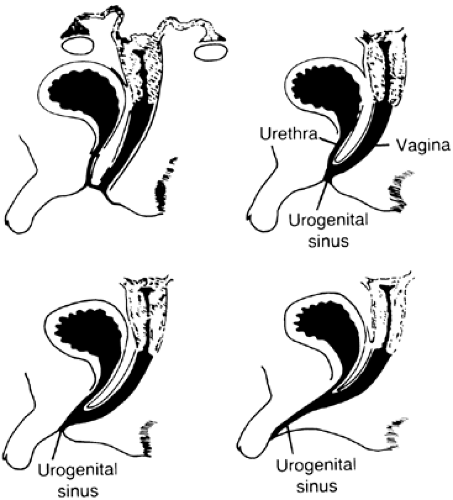 FIGURE 77-3. Variations in the differentiation of the external genitalia in congenital adrenal hyperplasia. In genetic females, adrenal androgen hypersecretion (enzymes 2, 4, and 5) is associated with various degrees of masculinization, leading to apparent male external genitalia. (From Grumbach MM, Ducharme J. The effects of androgens on fetal development: androgen-induced female pseudohermaphroditism. Fertil Steril 1960; 11:157.) |
Patients affected with the milder, nonclassic form of 21-hydroxylase deficiency may have signs of postnatal androgen excess.6 Except for rare cases showing mild clitoromegaly,
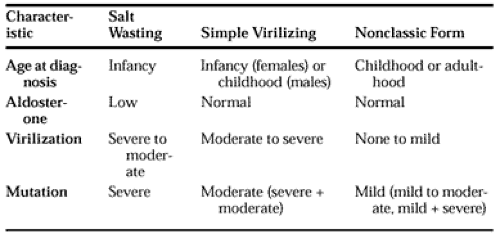
females with the nonclassic disorder are born with normal external genitalia. The syndrome of polycystic ovarian disease has often been confused with nonclassic CAH 21-hydroxylase deficiency in young women with hirsutism, oligomenorrhea, and diminished fertility. Precise clinical distinction between the classic simple virilizing disease and nonclassic disorder is sometimes difficult among males, because the hormonal reference standards for diagnosis represent a continuum. Moreover, because males do not manifest ambiguous genitalia as a sign of in utero androgen excess, the only other distinguishing clinical parameters are bone age and somatic growth pattern, which are nonpathognomonic. Phenotypic severity in nonclassic 21-hydroxylase deficiency varies greatly, and in some individuals the disease has been detected solely on the basis of hormonal or genetic testing in the course of family studies. Aldosterone synthesis is normal in patients with nonclassic 21-hydroxylase deficiency. Table 77-2 describes features distinguishing salt-wasting, simple virilizing, and nonclassic forms of 21-hydroxylase deficiency. Table 77-3 describes specific mutations.
females with the nonclassic disorder are born with normal external genitalia. The syndrome of polycystic ovarian disease has often been confused with nonclassic CAH 21-hydroxylase deficiency in young women with hirsutism, oligomenorrhea, and diminished fertility. Precise clinical distinction between the classic simple virilizing disease and nonclassic disorder is sometimes difficult among males, because the hormonal reference standards for diagnosis represent a continuum. Moreover, because males do not manifest ambiguous genitalia as a sign of in utero androgen excess, the only other distinguishing clinical parameters are bone age and somatic growth pattern, which are nonpathognomonic. Phenotypic severity in nonclassic 21-hydroxylase deficiency varies greatly, and in some individuals the disease has been detected solely on the basis of hormonal or genetic testing in the course of family studies. Aldosterone synthesis is normal in patients with nonclassic 21-hydroxylase deficiency. Table 77-2 describes features distinguishing salt-wasting, simple virilizing, and nonclassic forms of 21-hydroxylase deficiency. Table 77-3 describes specific mutations.
 |
 |
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


